

Digenic mutations in ALDH2 and ADH5 impair formaldehyde clearance and cause a multisystem disorder, AMeD syndrome
- PMID: 33355142
- PMCID: PMC11206199
- DOI: 10.1126/sciadv.abd7197
Digenic mutations in ALDH2 and ADH5 impair formaldehyde clearance and cause a multisystem disorder, AMeD syndrome
Abstract
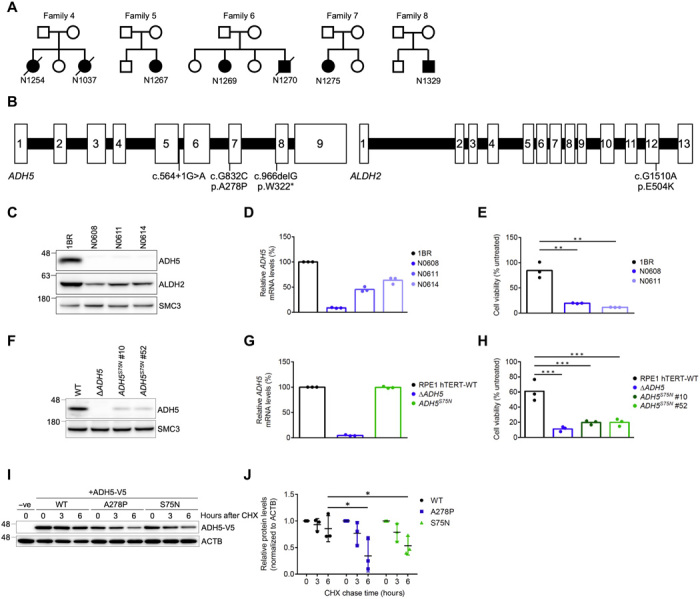
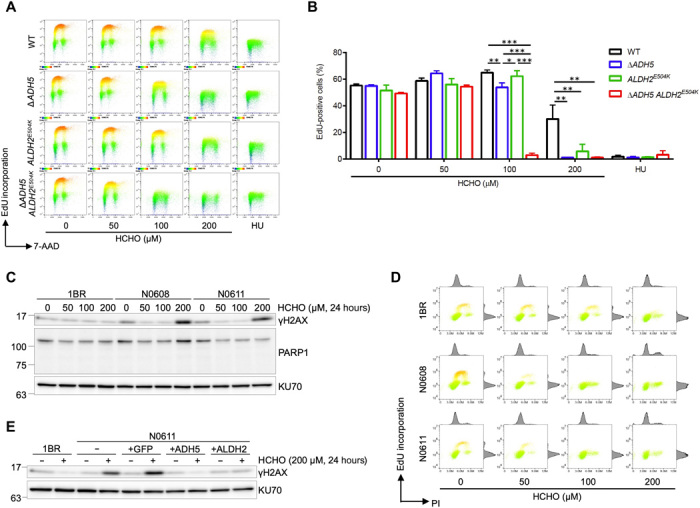
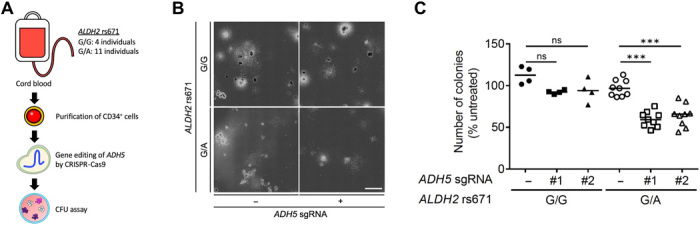
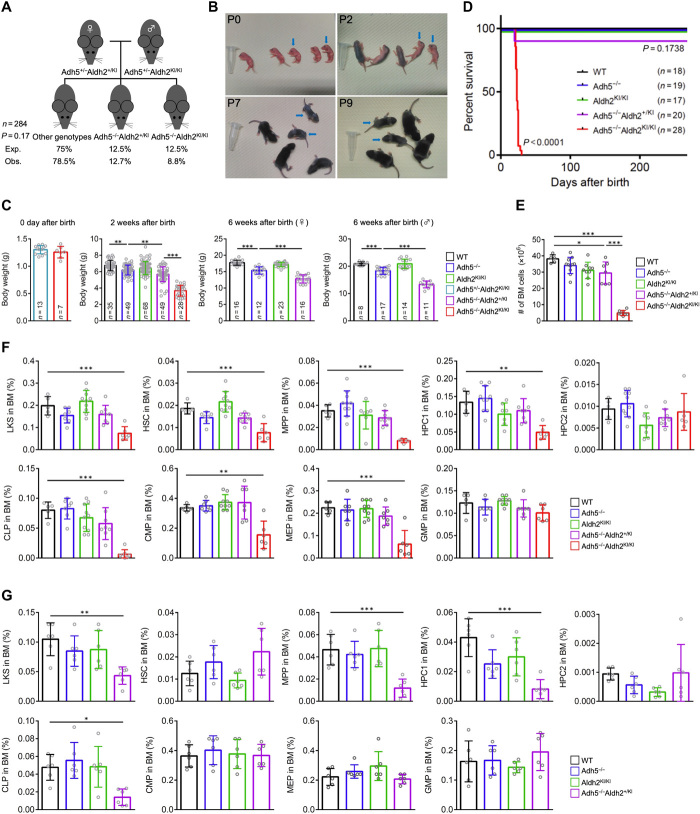
Rs671 in the aldehyde dehydrogenase 2 gene (ALDH2) is the cause of Asian alcohol flushing response after drinking. ALDH2 detoxifies endogenous aldehydes, which are the major source of DNA damage repaired by the Fanconi anemia pathway. Here, we show that the rs671 defective allele in combination with mutations in the alcohol dehydrogenase 5 gene, which encodes formaldehyde dehydrogenase (ADH5FDH ), causes a previously unidentified disorder, AMeD (aplastic anemia, mental retardation, and dwarfism) syndrome. Cellular studies revealed that a decrease in the formaldehyde tolerance underlies a loss of differentiation and proliferation capacity of hematopoietic stem cells. Moreover, Adh5-/-Aldh2 E506K/E506K double-deficient mice recapitulated key clinical features of AMeDS, showing short life span, dwarfism, and hematopoietic failure. Collectively, our results suggest that the combined deficiency of formaldehyde clearance mechanisms leads to the complex clinical features due to overload of formaldehyde-induced DNA damage, thereby saturation of DNA repair processes.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Burgos-Barragan G., Wit N., Meiser J., Dingler F. A., Pietzke M., Mulderrig L., Pontel L. B., Rosado I. V., Brewer T. F., Cordell R. L., Monks P. S., Chang C. J., Vazquez A., Patel K. J., Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548, 549–554 (2017). - PMC - PubMed
-
- Walport L. J., Hopkinson R. J., Schofield C. J., Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 16, 525–534 (2012). - PubMed
-
- Harada S., Agarwal D. P., Goedde H. W., Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. Lancet 2, 982 (1981). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
