

SPEG binds with desmin and its deficiency causes defects in triad and focal adhesion proteins
- PMID: 33355670
- PMCID: PMC8485222
- DOI: 10.1093/hmg/ddaa276
SPEG binds with desmin and its deficiency causes defects in triad and focal adhesion proteins
Abstract
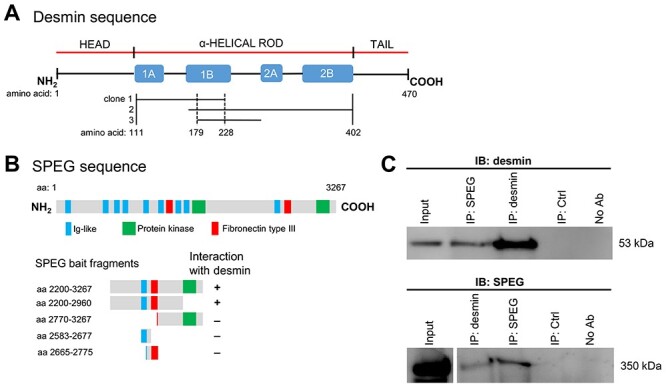
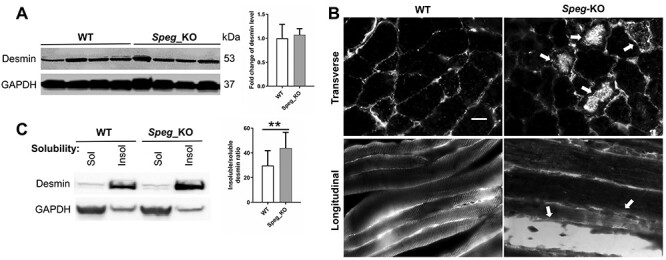
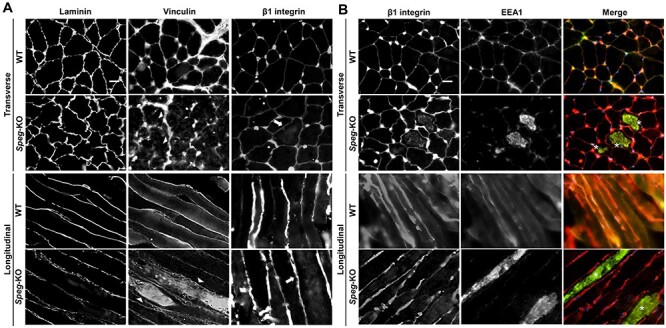
Striated preferentially expressed gene (SPEG), a member of the myosin light chain kinase family, is localized at the level of triad surrounding myofibrils in skeletal muscles. In humans, SPEG mutations are associated with centronuclear myopathy and cardiomyopathy. Using a striated muscle-specific Speg-knockout (KO) mouse model, we have previously shown that SPEG is critical for triad maintenance and calcium handling. Here, we further examined the molecular function of SPEG and characterized the effects of SPEG deficiency on triad and focal adhesion proteins. We used yeast two-hybrid assay, and identified desmin, an intermediate filament protein, to interact with SPEG and confirmed this interaction by co-immunoprecipitation. Using domain-mapping assay, we defined that Ig-like and fibronectin III domains of SPEG interact with rod domain of desmin. In skeletal muscles, SPEG depletion leads to desmin aggregates in vivo and a shift in desmin equilibrium from soluble to insoluble fraction. We also profiled the expression and localization of triadic proteins in Speg-KO mice using western blot and immunofluorescence. The amount of RyR1 and triadin were markedly reduced, whereas DHPRα1, SERCA1 and triadin were abnormally accumulated in discrete areas of Speg-KO myofibers. In addition, Speg-KO muscles exhibited internalized vinculin and β1 integrin, both of which are critical components of the focal adhesion complex. Further, β1 integrin was abnormally accumulated in early endosomes of Speg-KO myofibers. These results demonstrate that SPEG-deficient skeletal muscles exhibit several pathological features similar to those seen in MTM1 deficiency. Defects of shared cellular pathways may underlie these structural and functional abnormalities in both types of diseases.
© The Author(s) 2020. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures


References
-
- Wilmshurst, J.M., Lillis, S., Zhou, H., Pillay, K., Henderson, H., Kress, W., Muller, C.R., Ndondo, A., Cloke, V., Cullup, T. et al. (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann. Neurol., 68, 717–726. - PubMed
-
- Nicot, A.S., Toussaint, A., Tosch, V., Kretz, C., Wallgren-Pettersson, C., Iwarsson, E., Kingston, H., Garnier, J.M., Biancalana, V., Oldfors, A. et al. (2007) Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet., 39, 1134–1139. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
