

Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ
- PMID: 33357446
- PMCID: PMC7836100
- DOI: 10.1016/j.cell.2020.12.021
Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ
Abstract
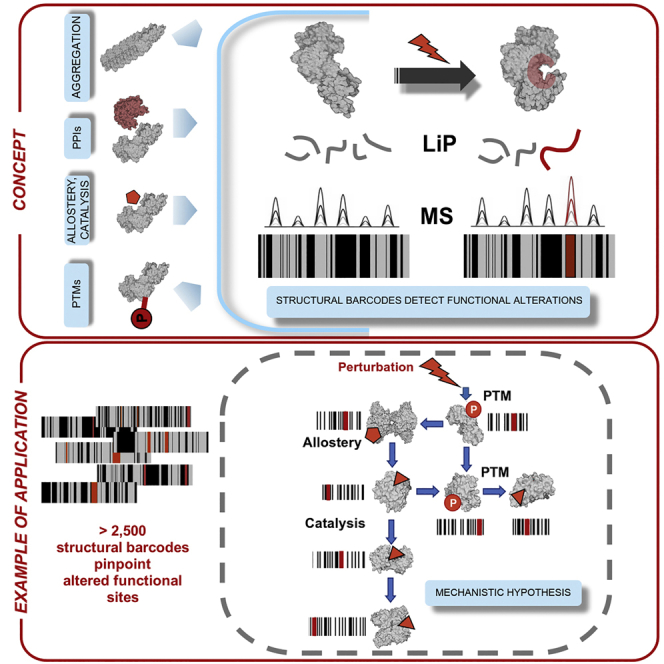
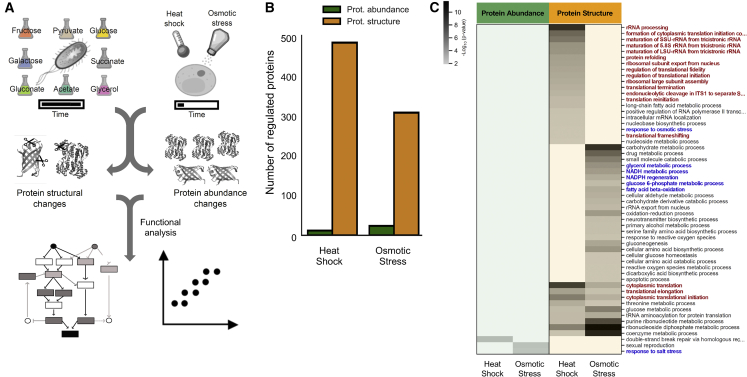
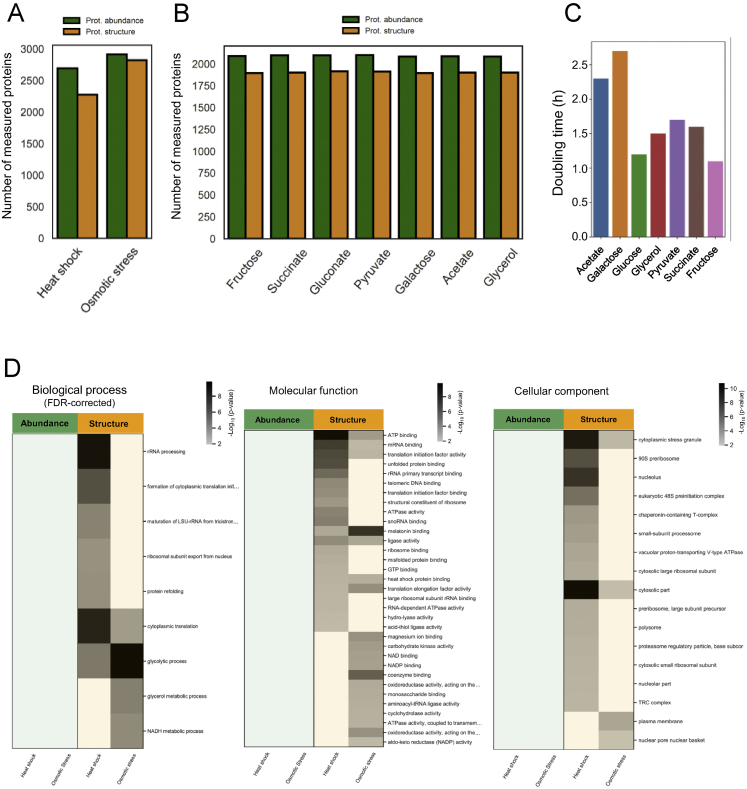
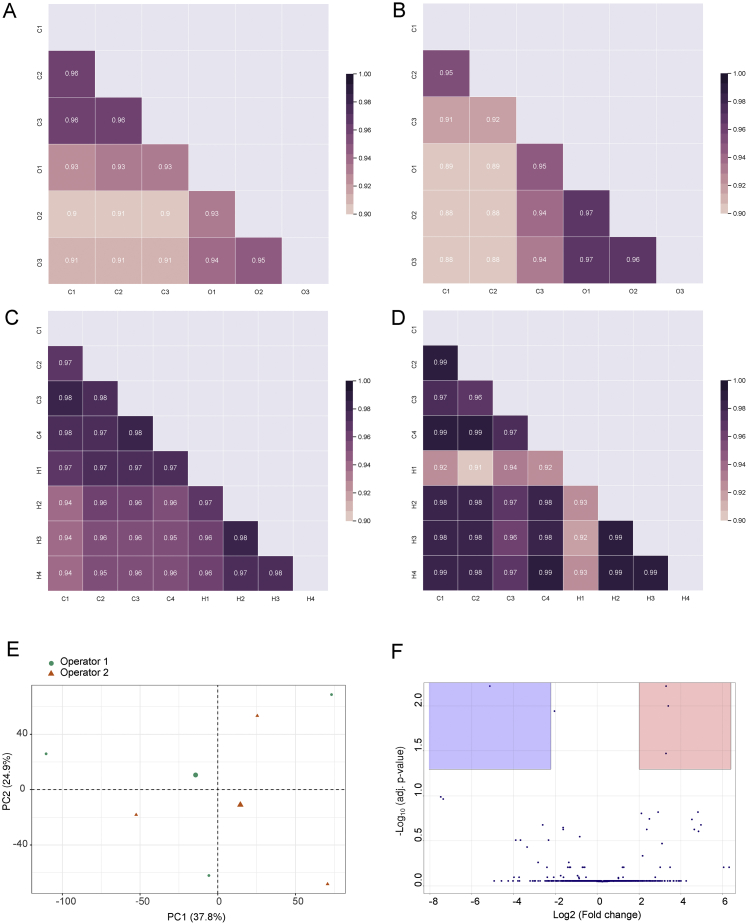
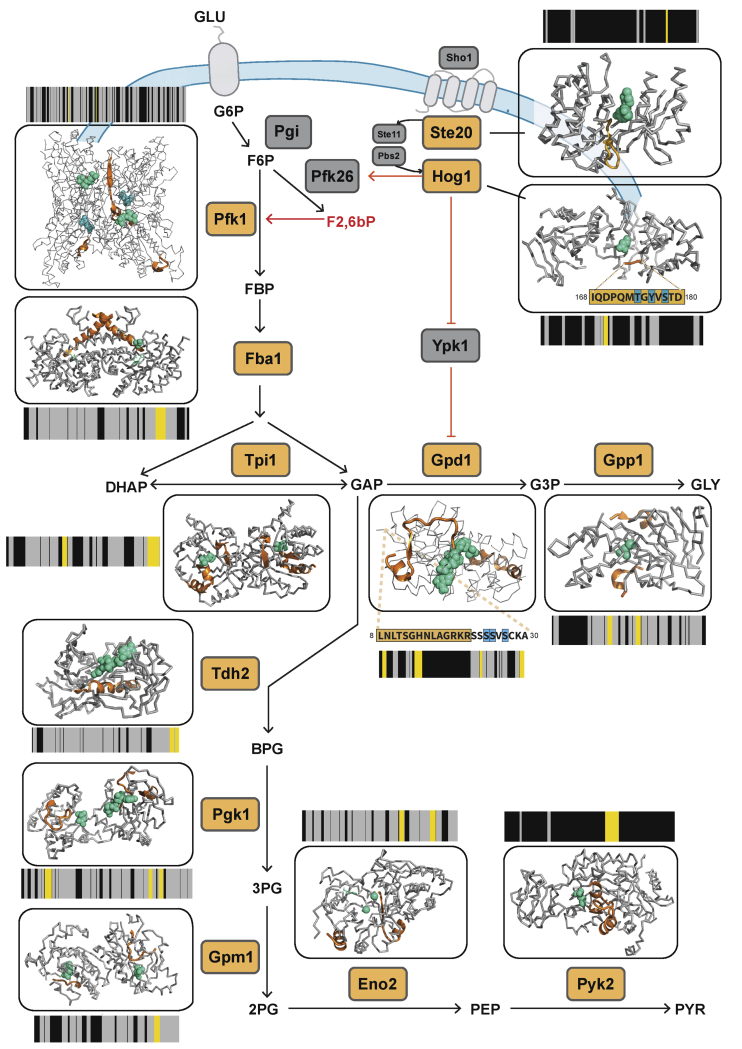
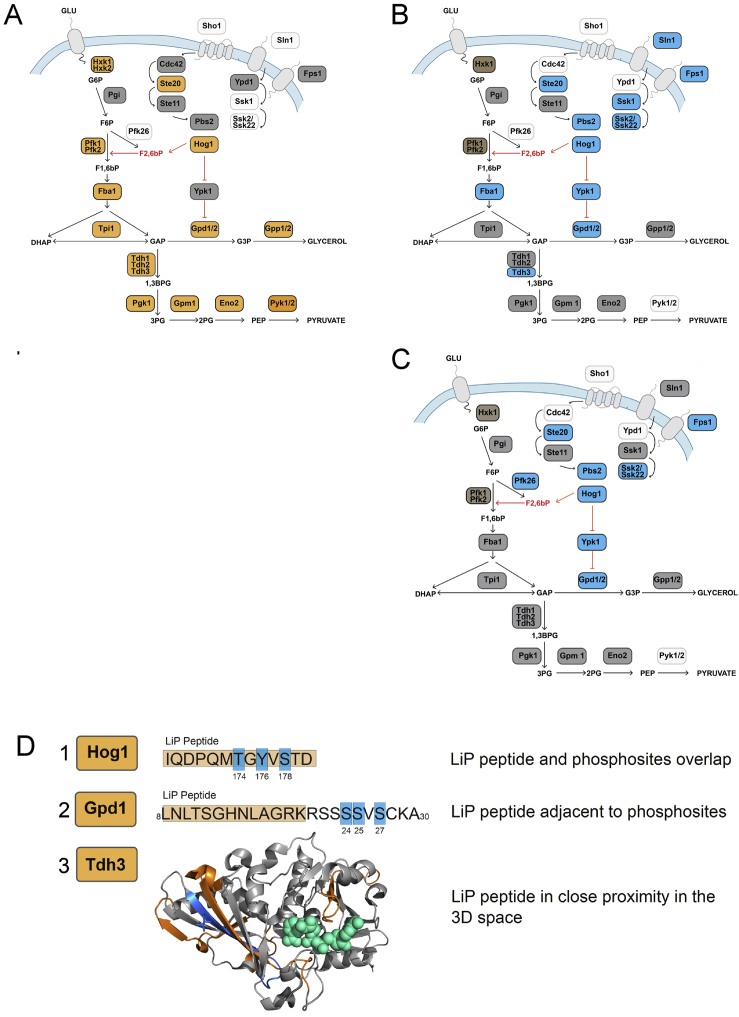
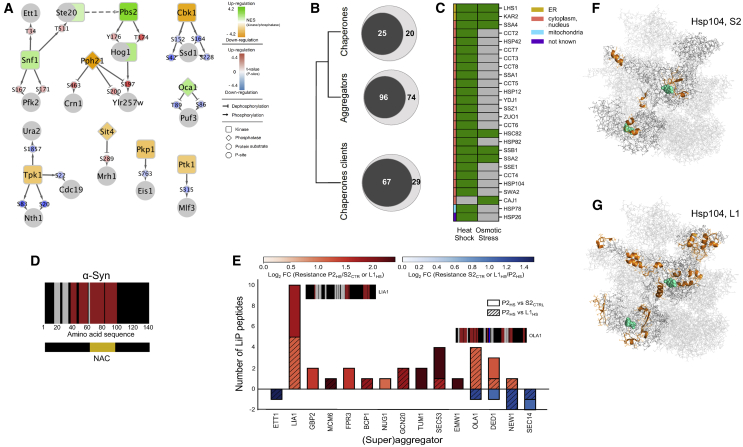
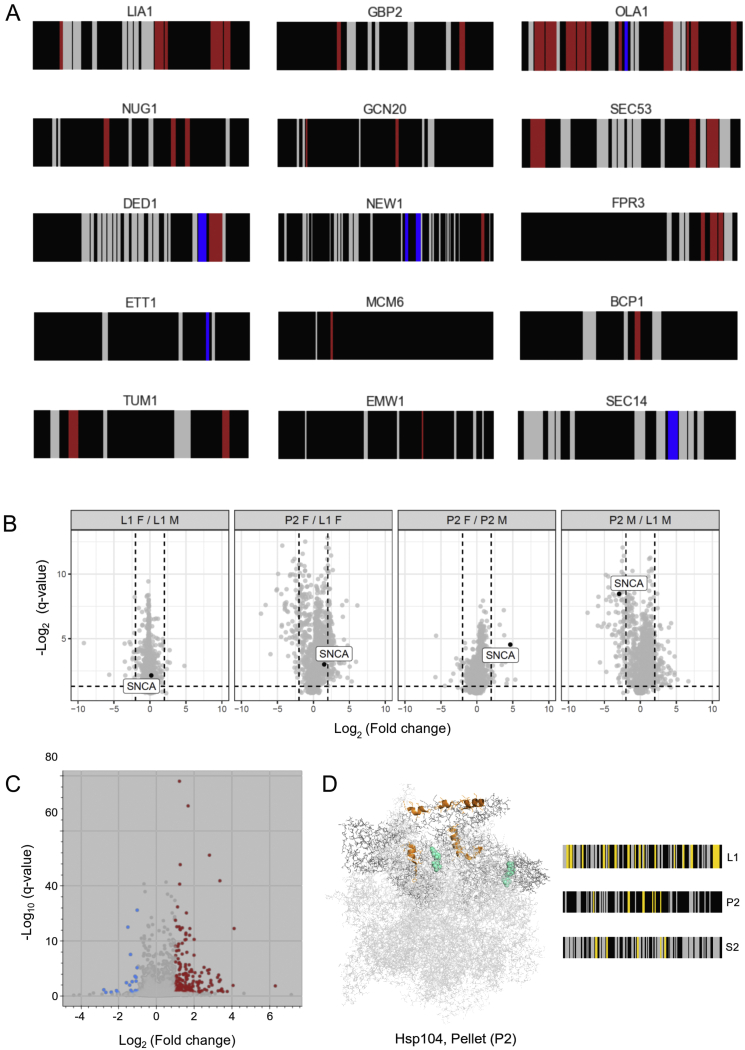
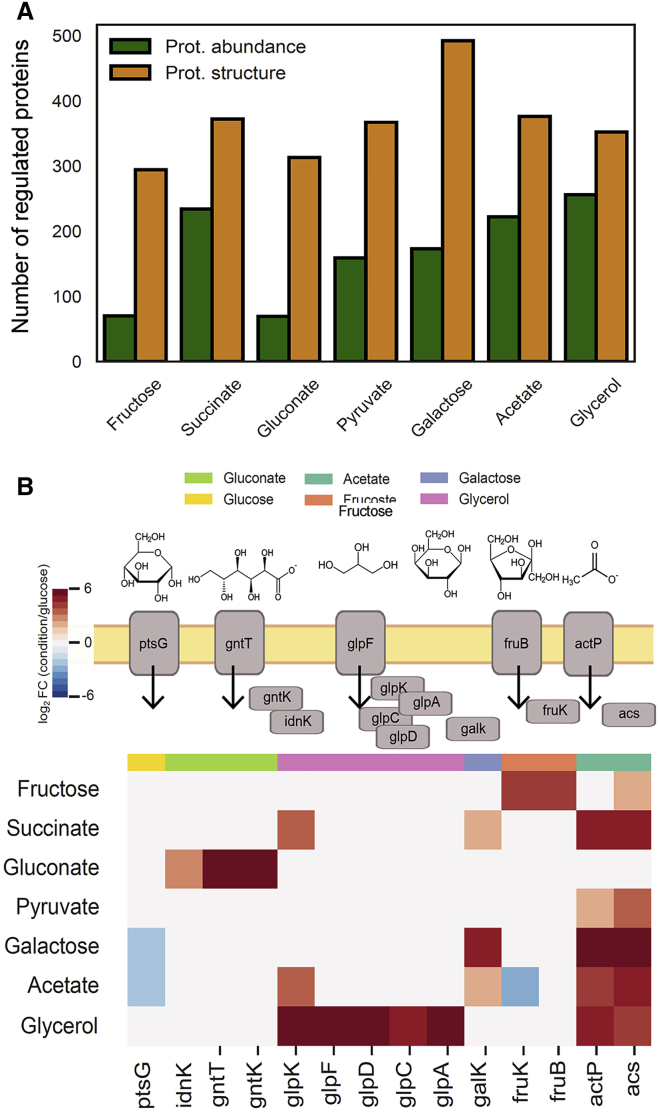
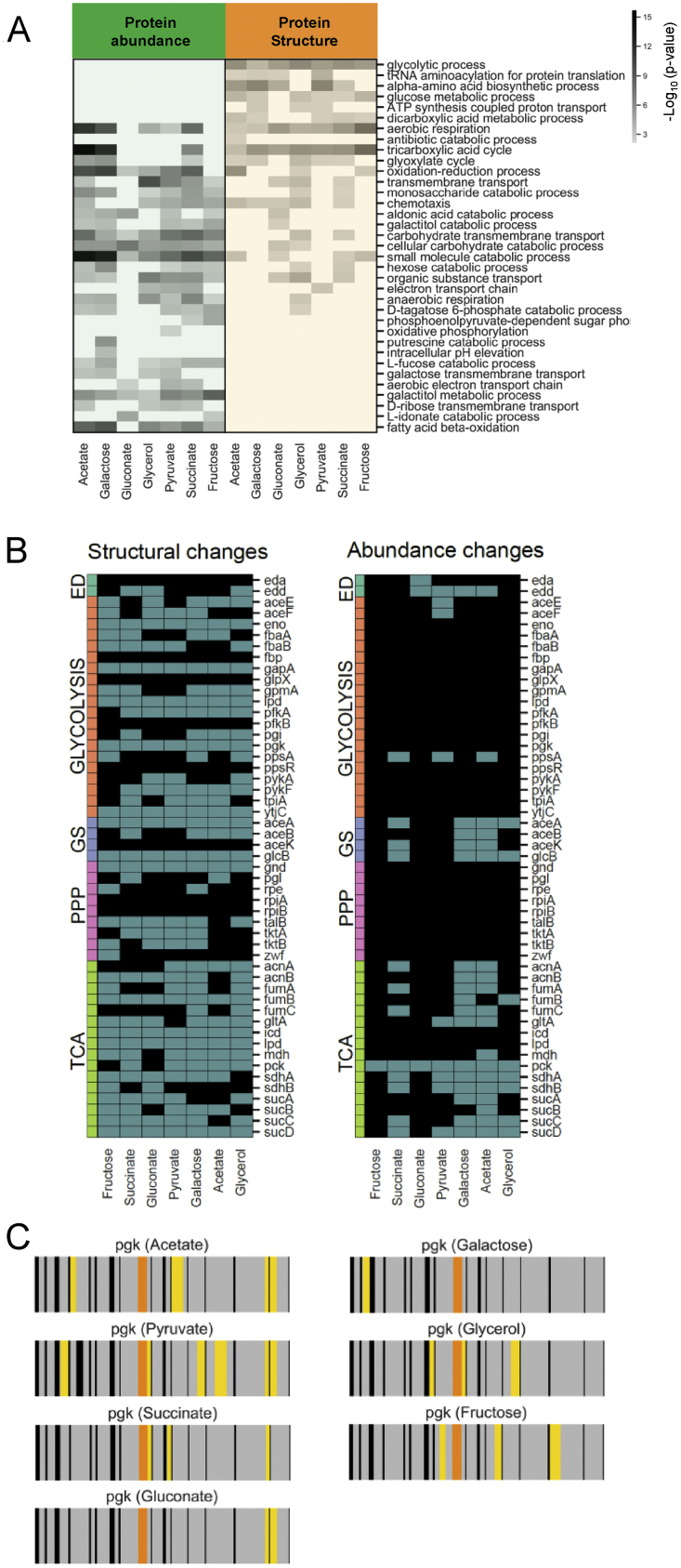
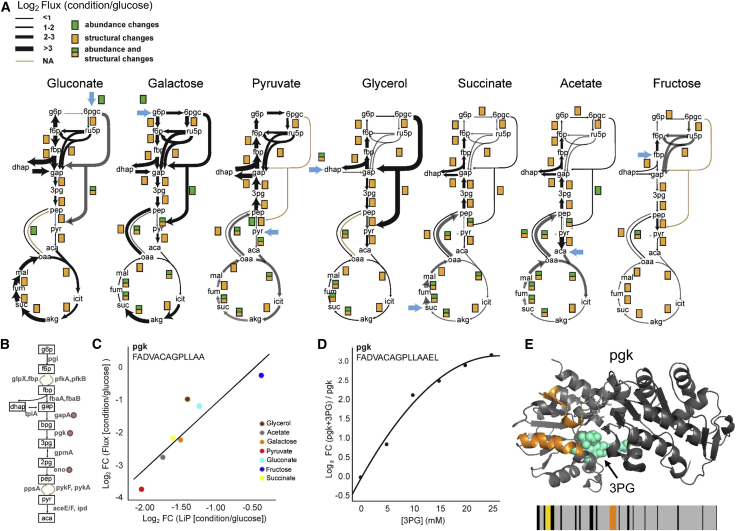
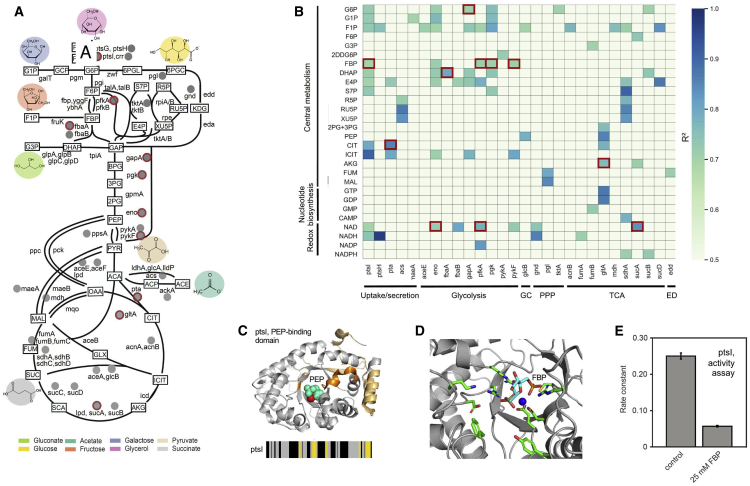
Biological processes are regulated by intermolecular interactions and chemical modifications that do not affect protein levels, thus escaping detection in classical proteomic screens. We demonstrate here that a global protein structural readout based on limited proteolysis-mass spectrometry (LiP-MS) detects many such functional alterations, simultaneously and in situ, in bacteria undergoing nutrient adaptation and in yeast responding to acute stress. The structural readout, visualized as structural barcodes, captured enzyme activity changes, phosphorylation, protein aggregation, and complex formation, with the resolution of individual regulated functional sites such as binding and active sites. Comparison with prior knowledge, including other 'omics data, showed that LiP-MS detects many known functional alterations within well-studied pathways. It suggested distinct metabolite-protein interactions and enabled identification of a fructose-1,6-bisphosphate-based regulatory mechanism of glucose uptake in E. coli. The structural readout dramatically increases classical proteomics coverage, generates mechanistic hypotheses, and paves the way for in situ structural systems biology.
Keywords: E. coli; functional proteomics; limited proteolysis; mass spectrometry; metabolism; protein aggregation; structural biology; structural proteomics; structural systems biology; yeast.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.P. is a scientific advisor for the company Biognosys AG (Zurich, Switzerland) and an inventor of a patent licensed by Biognosys AG that covers the LiP-MS method used in this manuscript.
Figures

Comment in
-
"Structuromics": another step toward a holistic view of the cell.Cell. 2021 Jan 21;184(2):301-303. doi: 10.1016/j.cell.2020.12.030. Cell. 2021. PMID: 33482097
-
Probing dynamic proteomes.Nat Methods. 2021 Feb;18(2):120. doi: 10.1038/s41592-021-01069-8. Nat Methods. 2021. PMID: 33542505 No abstract available.
References
-
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. - PubMed
-
- Alexa A., Rahnenführer J., Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
