

A new FRET-based platform to track substrate ubiquitination by fluorescence
- PMID: 33361156
- PMCID: PMC7948536
- DOI: 10.1074/jbc.RA120.016858
A new FRET-based platform to track substrate ubiquitination by fluorescence
Abstract
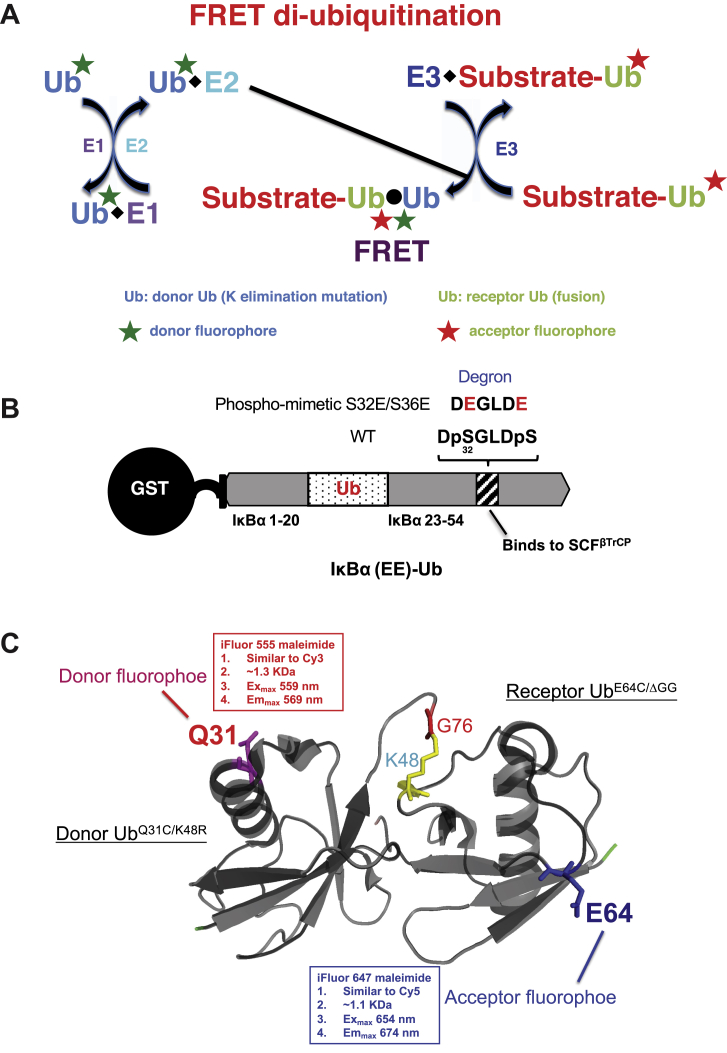
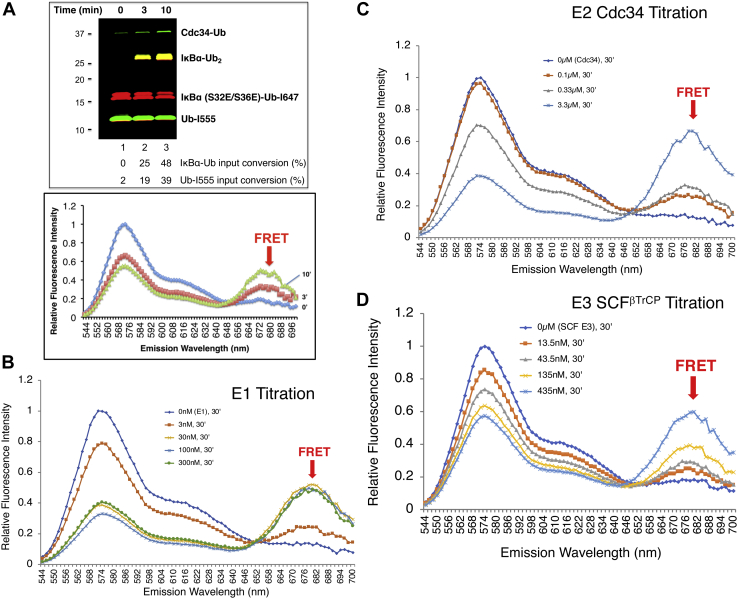
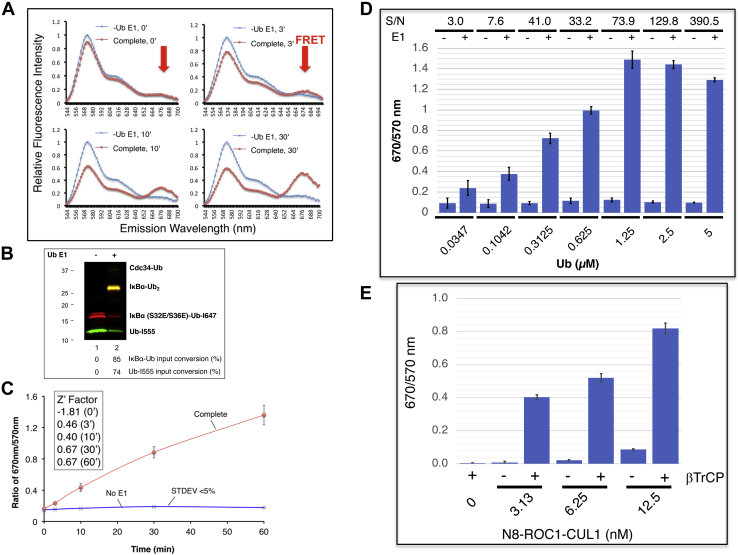
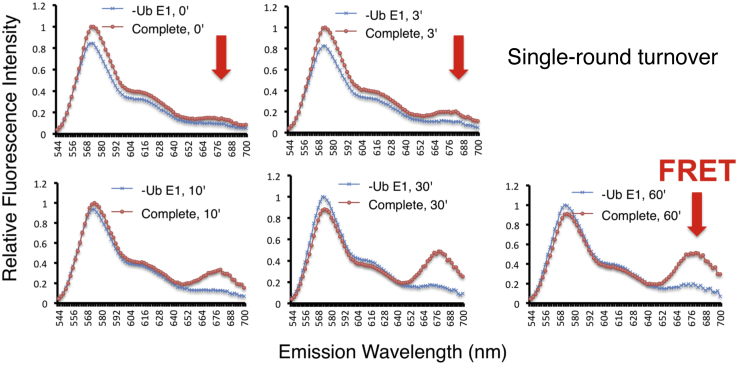
Post-translational modification of protein by ubiquitin (Ub) alters the stability, subcellular location, or function of the target protein, thereby impacting numerous biological processes and directly contributing to myriad cellular defects or disease states, such as cancer. Tracking substrate ubiquitination by fluorescence provides opportunities for advanced reaction dynamics studies and for translational research including drug discovery. However, fluorescence-based techniques in ubiquitination studies remain underexplored at least partly because of challenges associated with Ub chain complexity and requirement for additional substrate modification. Here we describe a general strategy, FRET diubiquitination, to track substrate ubiquitination by fluorescence. This platform produces a uniform di-Ub product depending on specific interactions between a substrate and its cognate E3 Ub ligase. The diubiquitination creates proximity between the Ub-linked donor and acceptor fluorophores, respectively, enabling energy transfer to yield a distinct fluorescent signal. FRET diubiquitination relies on Ub-substrate fusion, which can be implemented using either one of the two validated strategies. Method 1 is the use of recombinant substrate-Ub fusion, applicable to all substrate peptides that can bind to E3. Method 2 is a chemoenzymatic ligation approach that employs synthetic chemistry to fuse Ub with a substrate peptide containing desired modification. Taken together, our new FRET-based diubiquitination system provides a timely technology of potential to advance both basic research and translation sciences.
Keywords: E3 ubiquitin ligase; FRET; chemoenzymatic ligation; kinetics; protein degradation; ubiquitination.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
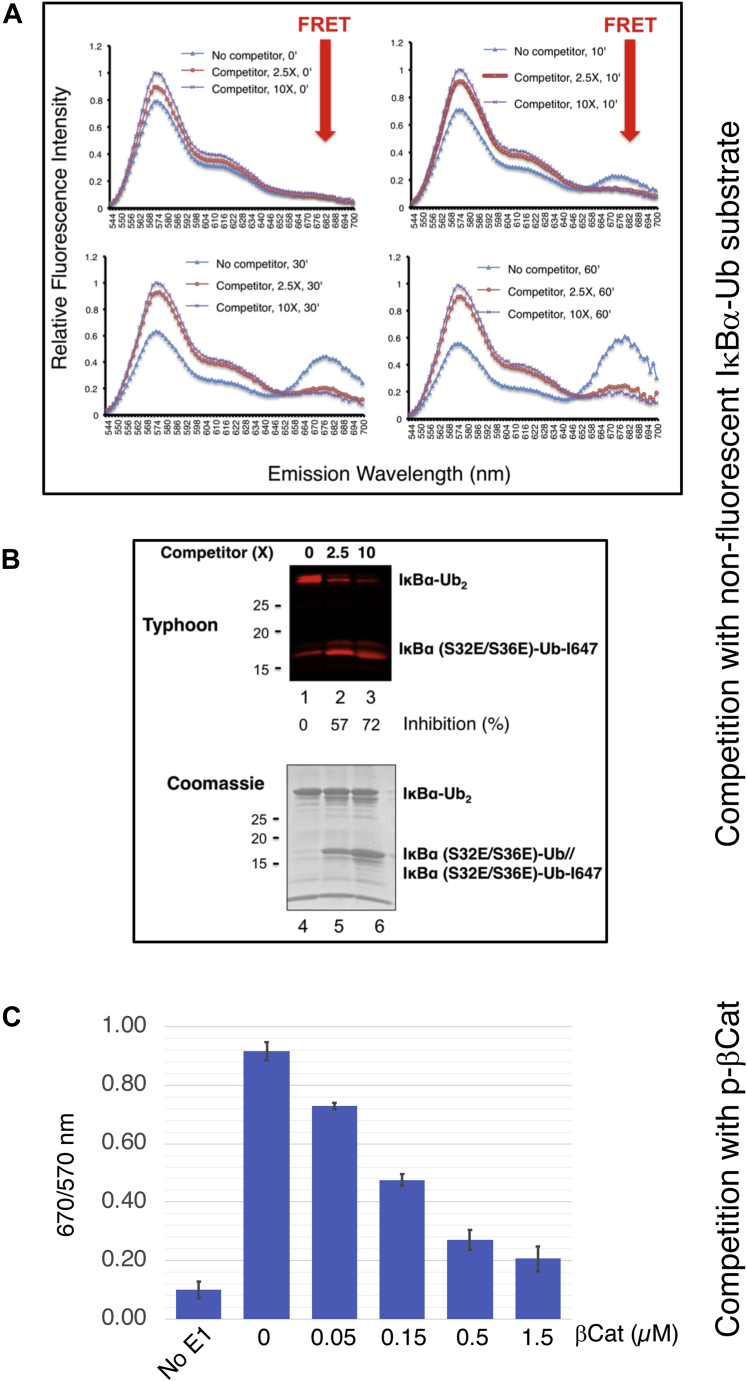
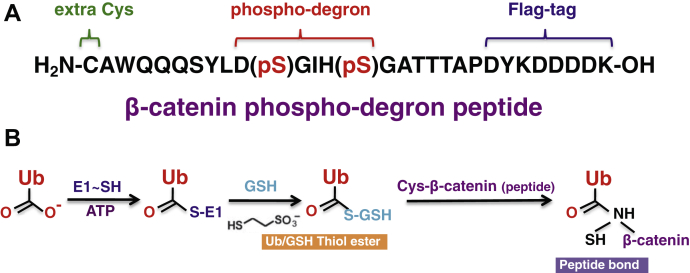
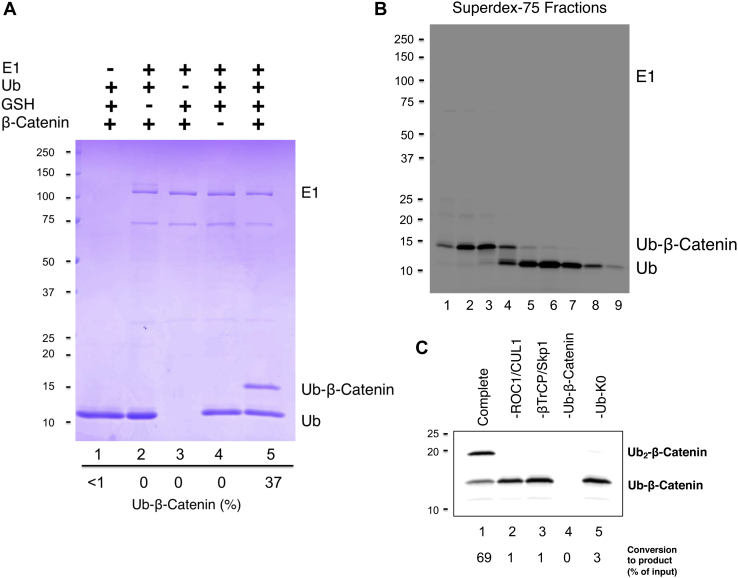
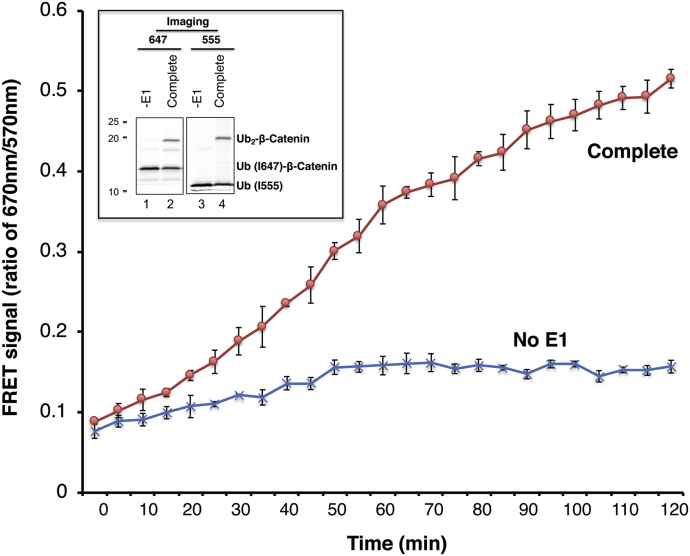
References
-
- Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. - PubMed
-
- Rajalingam K., Dikic I. SnapShot: expanding the ubiquitin code. Cell. 2016;164:1074–1074.e1. - PubMed
-
- Morreale F.E., Walden H. Types of ubiquitin ligases. Cell. 2016;165:248–248.e1. - PubMed
-
- Skaar J.R., Pagan J.K., Pagano M. SnapShot: F box proteins I. Cell. 2009;137:1160–1160.e1. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
