

DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes
- PMID: 33361332
- PMCID: PMC7812826
- DOI: 10.1073/pnas.2017525118
DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes
Abstract
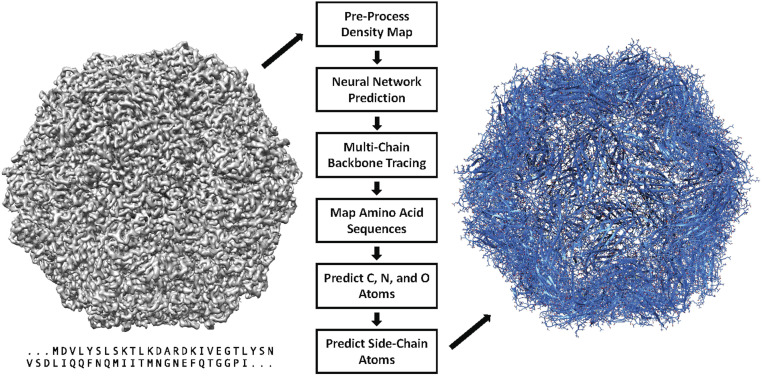
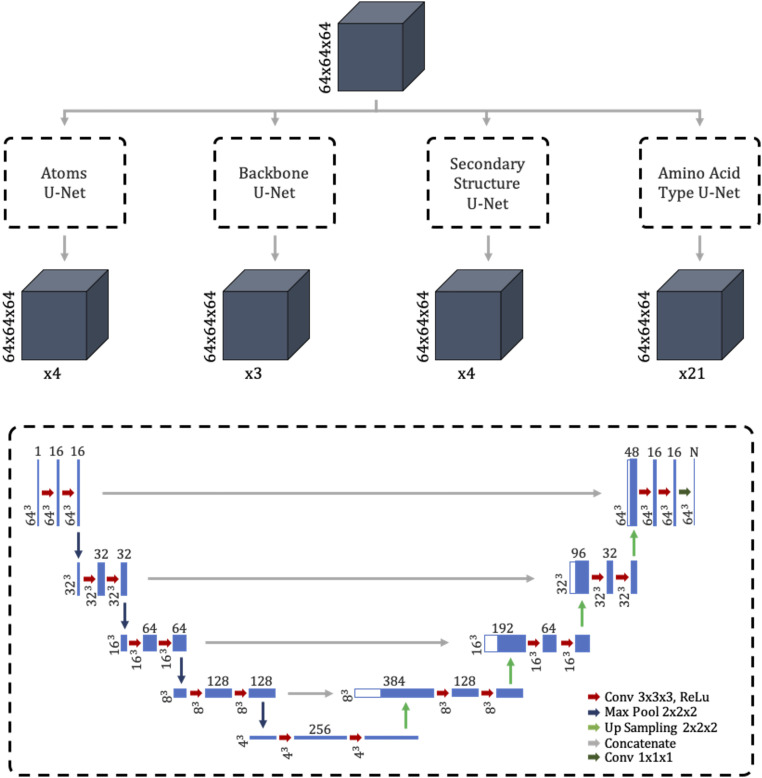
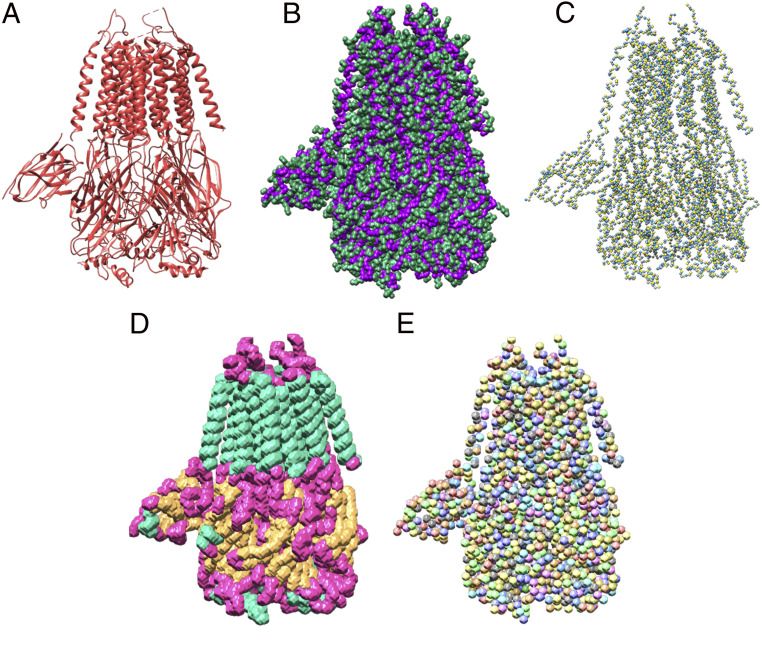
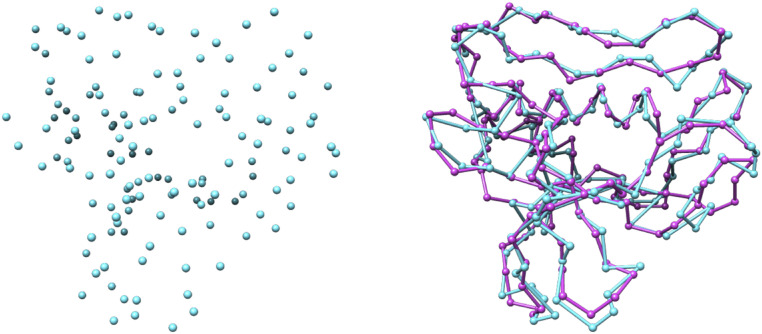
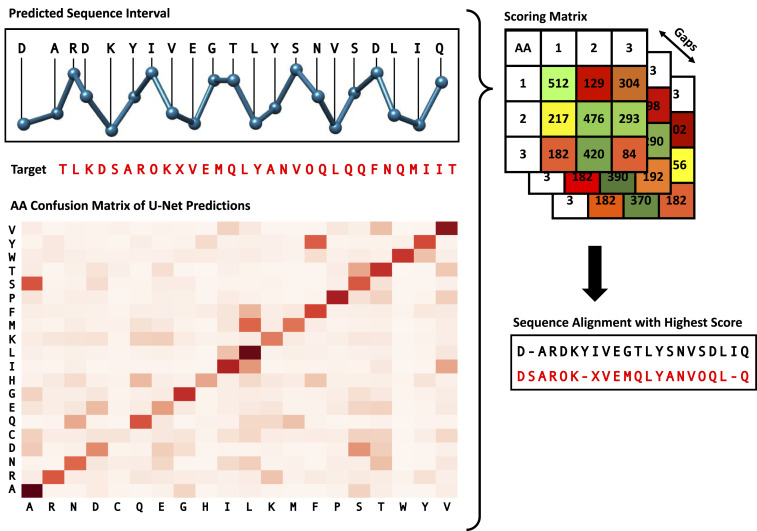
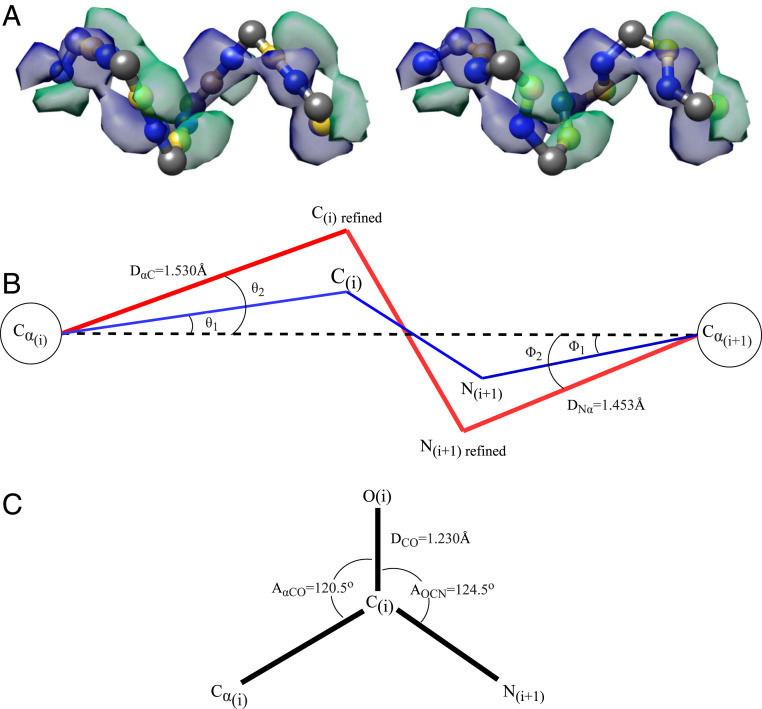
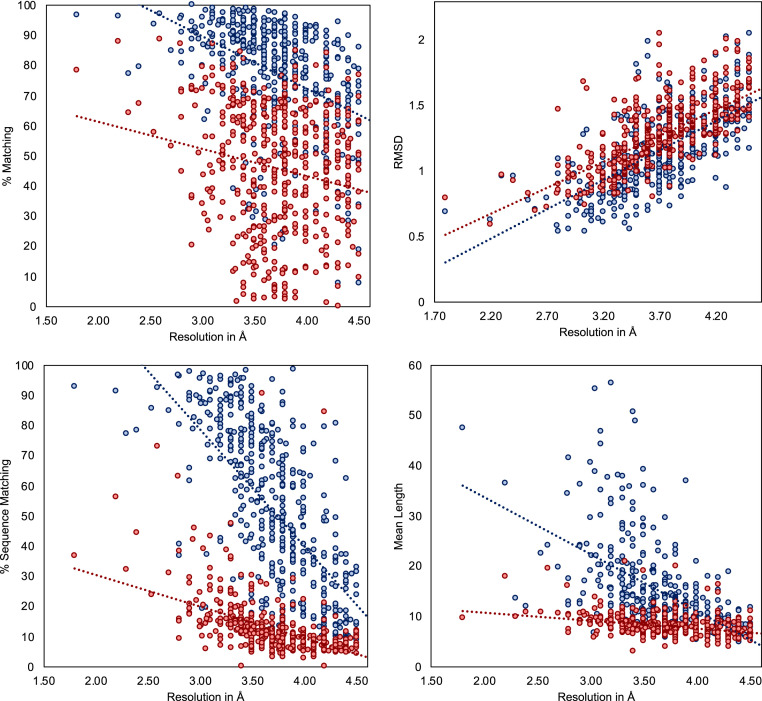
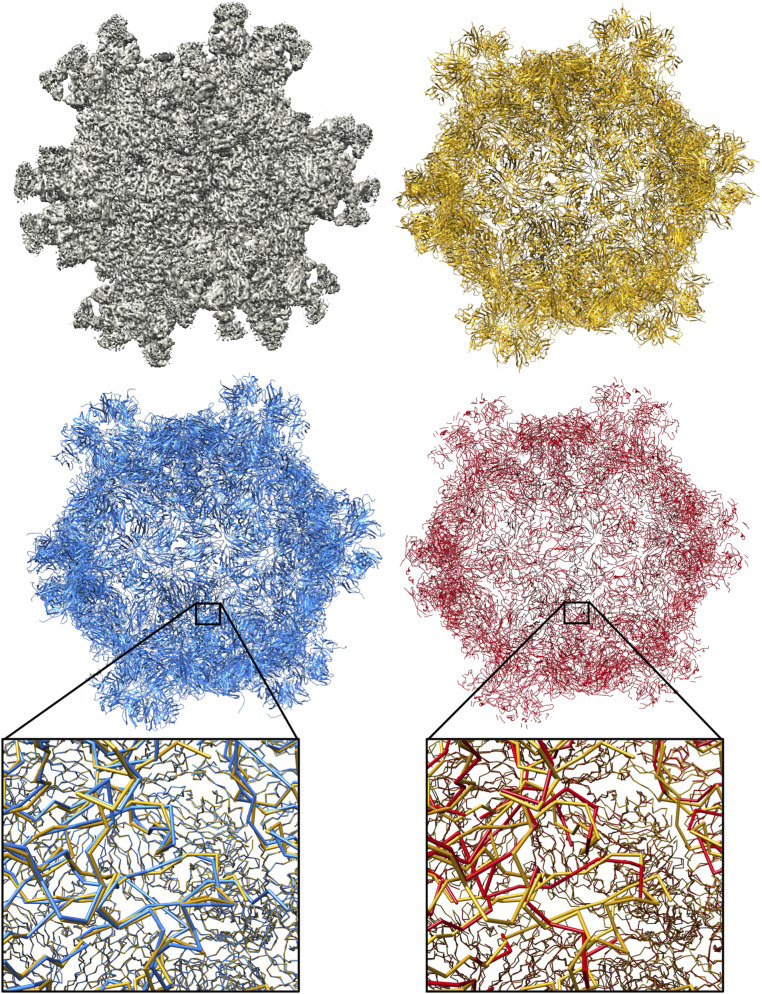
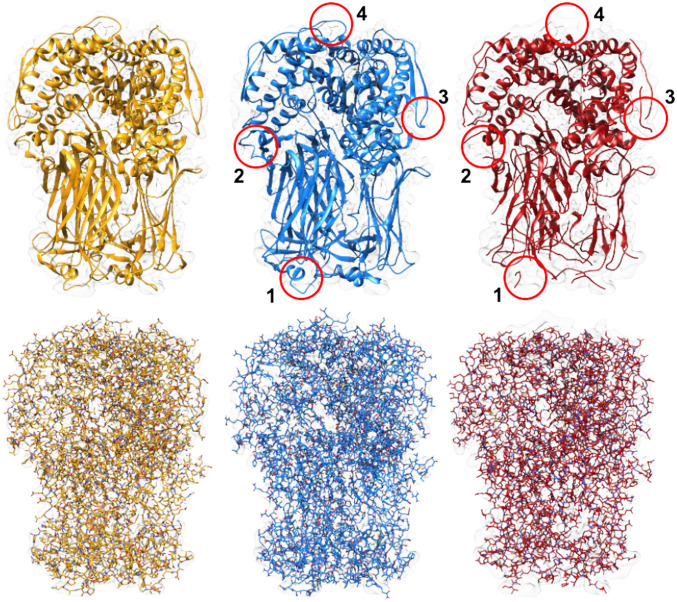
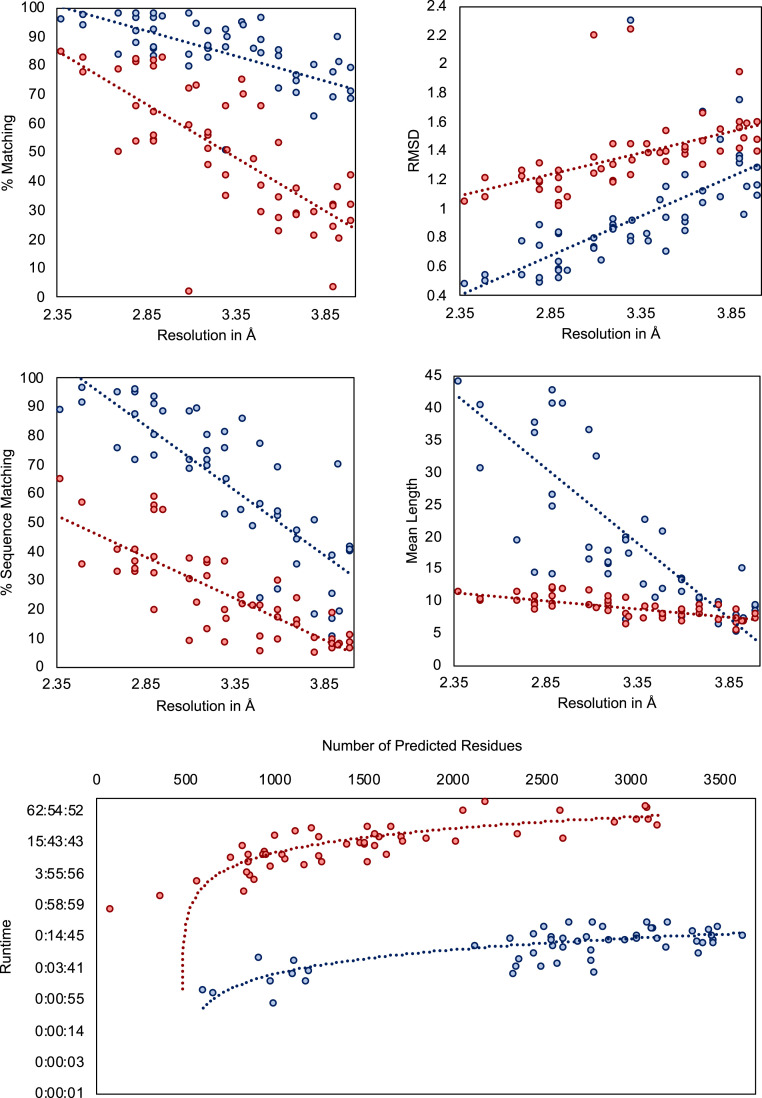
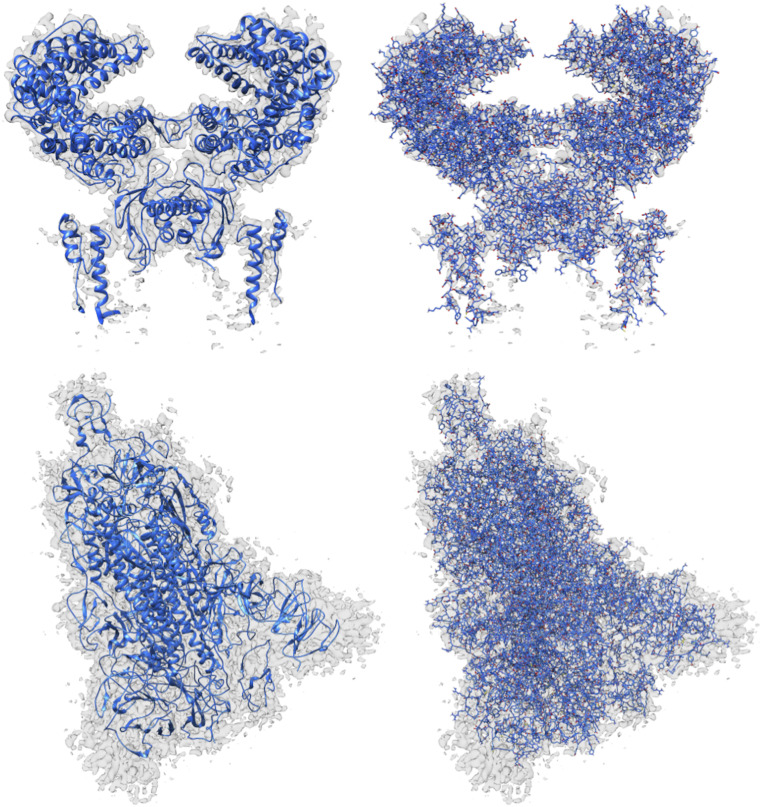
Information about macromolecular structure of protein complexes and related cellular and molecular mechanisms can assist the search for vaccines and drug development processes. To obtain such structural information, we present DeepTracer, a fully automated deep learning-based method for fast de novo multichain protein complex structure determination from high-resolution cryoelectron microscopy (cryo-EM) maps. We applied DeepTracer on a previously published set of 476 raw experimental cryo-EM maps and compared the results with a current state of the art method. The residue coverage increased by over 30% using DeepTracer, and the rmsd value improved from 1.29 Å to 1.18 Å. Additionally, we applied DeepTracer on a set of 62 coronavirus-related cryo-EM maps, among them 10 with no deposited structure available in EMDataResource. We observed an average residue match of 84% with the deposited structures and an average rmsd of 0.93 Å. Additional tests with related methods further exemplify DeepTracer's competitive accuracy and efficiency of structure modeling. DeepTracer allows for exceptionally fast computations, making it possible to trace around 60,000 residues in 350 chains within only 2 h. The web service is globally accessible at https://deeptracer.uw.edu.
Keywords: complex; cryo-EM; de novo; modeling; structure.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures

References
-
- Branden C. I., Tooze J., Introduction to Protein Structure (Garland Science, 2012).
-
- Callaway E., Revolutionary cryo-EM is taking over structural biology. Nature 578, 201 (2020). - PubMed
-
- Bai X. C., McMullan G., Scheres S. H., How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 (2015). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
