

Detailed genetic and functional analysis of the hDMDdel52/mdx mouse model
- PMID: 33362201
- PMCID: PMC7757897
- DOI: 10.1371/journal.pone.0244215
Detailed genetic and functional analysis of the hDMDdel52/mdx mouse model
Abstract
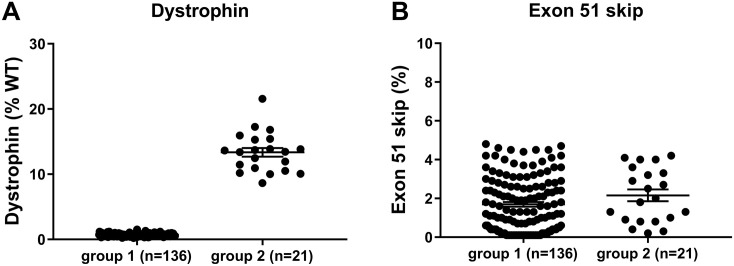
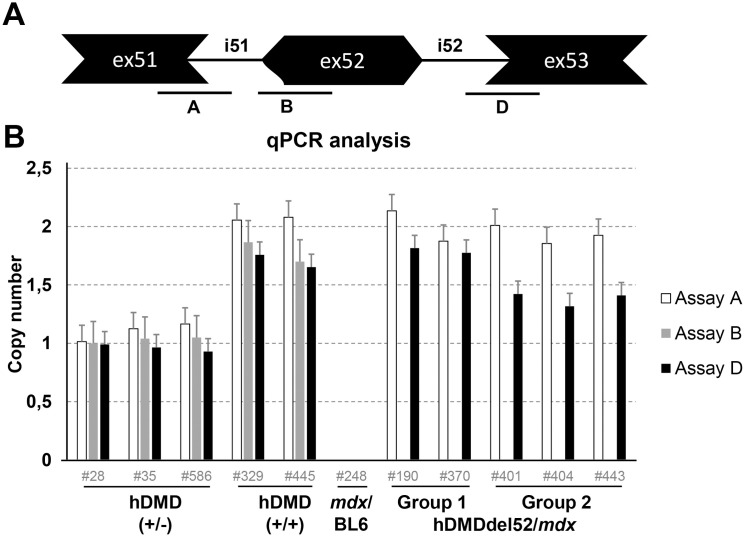
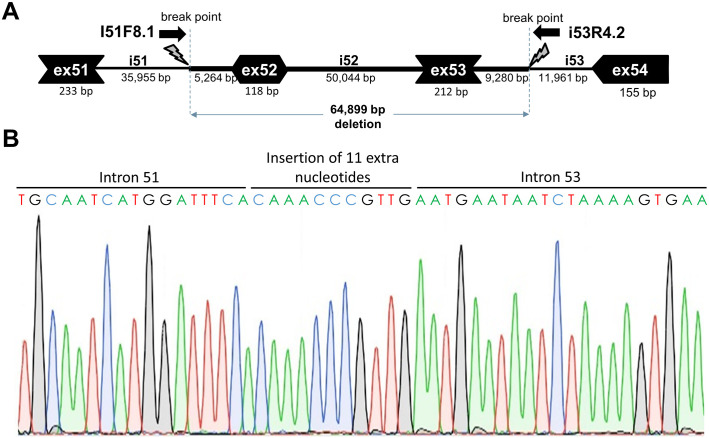
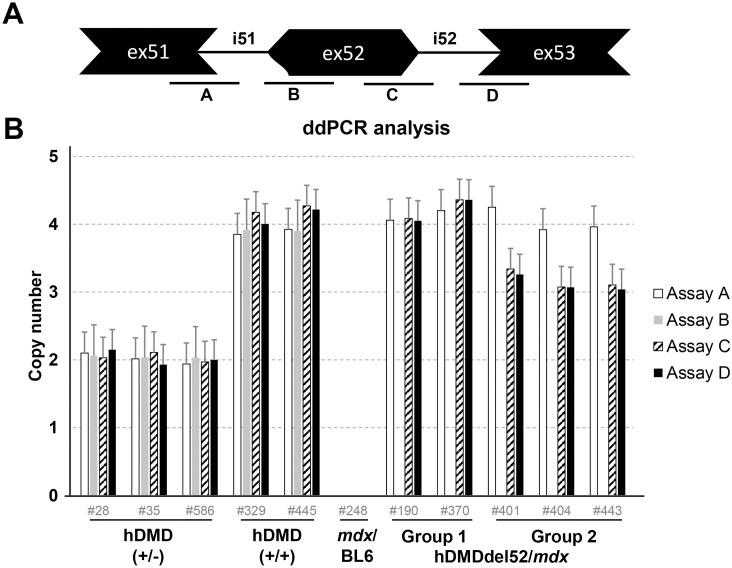
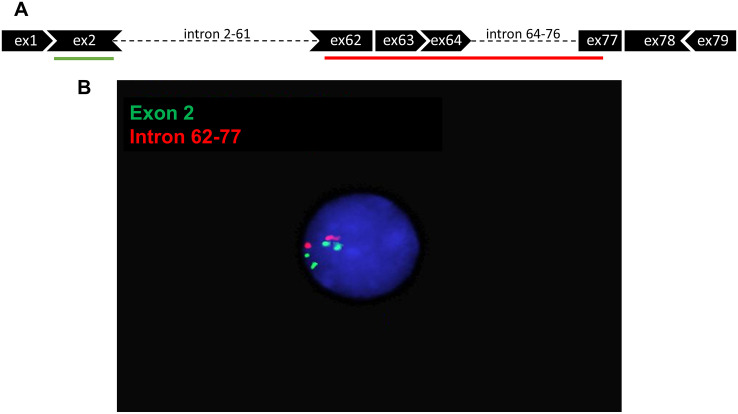
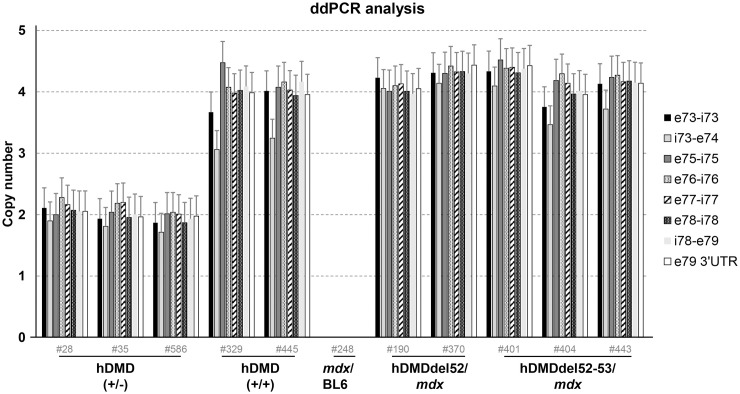
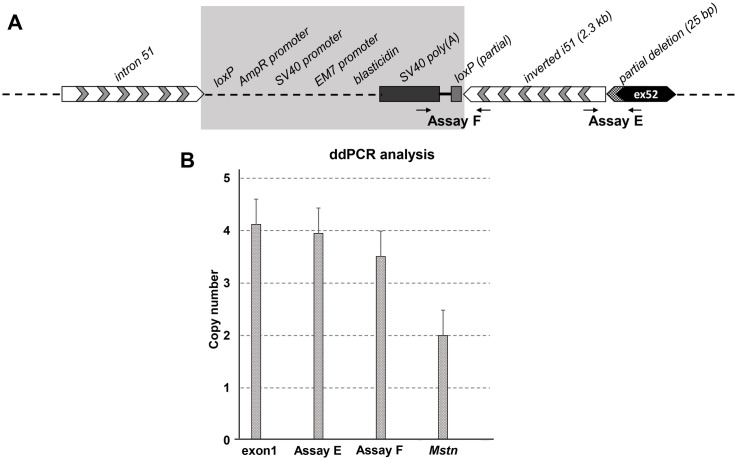
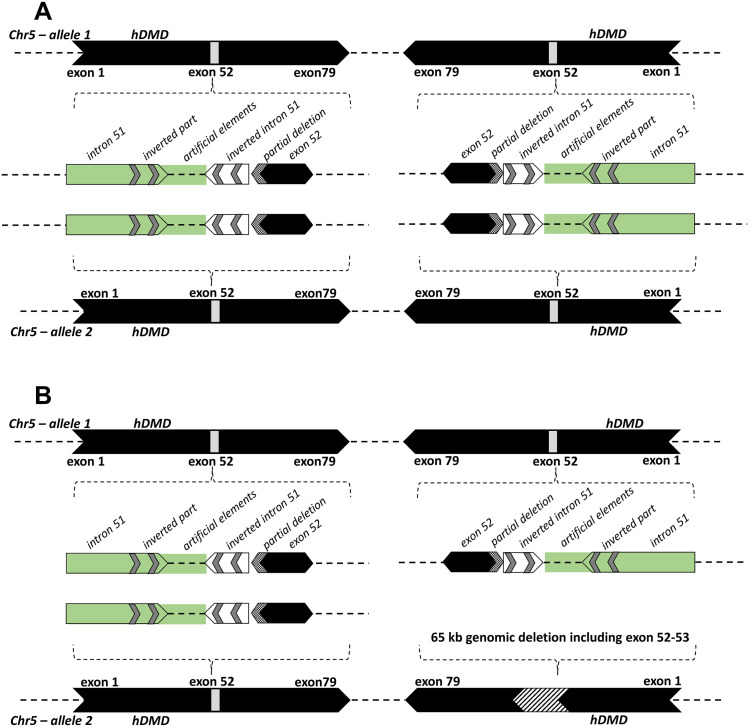
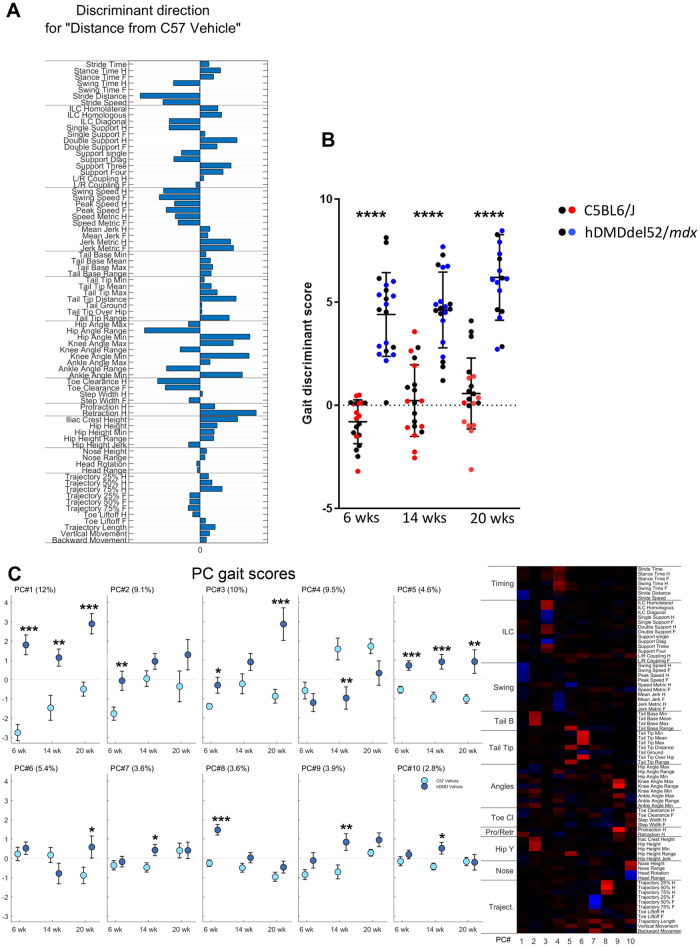
Duchenne muscular dystrophy (DMD) is a severe, progressive neuromuscular disorder caused by reading frame disrupting mutations in the DMD gene leading to absence of functional dystrophin. Antisense oligonucleotide (AON)-mediated exon skipping is a therapeutic approach aimed at restoring the reading frame at the pre-mRNA level, allowing the production of internally truncated partly functional dystrophin proteins. AONs work in a sequence specific manner, which warrants generating humanized mouse models for preclinical tests. To address this, we previously generated the hDMDdel52/mdx mouse model using transcription activator like effector nuclease (TALEN) technology. This model contains mutated murine and human DMD genes, and therefore lacks mouse and human dystrophin resulting in a dystrophic phenotype. It allows preclinical evaluation of AONs inducing the skipping of human DMD exons 51 and 53 and resulting in restoration of dystrophin synthesis. Here, we have further characterized this model genetically and functionally. We discovered that the hDMD and hDMDdel52 transgene is present twice per locus, in a tail-to-tail-orientation. Long-read sequencing revealed a partial deletion of exon 52 (first 25 bp), and a 2.3 kb inversion in intron 51 in both copies. These new findings on the genomic make-up of the hDMD and hDMDdel52 transgene do not affect exon 51 and/or 53 skipping, but do underline the need for extensive genetic analysis of mice generated with genome editing techniques to elucidate additional genetic changes that might have occurred. The hDMDdel52/mdx mice were also evaluated functionally using kinematic gait analysis. This revealed a clear and highly significant difference in overall gait between hDMDdel52/mdx mice and C57BL6/J controls. The motor deficit detected in the model confirms its suitability for preclinical testing of exon skipping AONs for human DMD at both the functional and molecular level.
Conflict of interest statement
AAR discloses being employed by LUMC which has patents on exon skipping technology, some of which has been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents AAR is entitled to a share of royalties. AAR further discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, CRISPR Therapeutics, Summit PLC, Alpha Anomeric, BioMarin Pharmaceuticals Inc., Eisai, Astra Zeneca, Santhera, Audentes, Global Guidepoint and GLG consultancy, Grunenthal, Wave and BioClinica, having been a member of the Duchenne Network Steering Committee (BioMarin) and being a member of the scientific advisory boards of ProQR, Sarepta, Silence and Philae Pharmaceuticals. Remuneration for these activities is paid to LUMC. LUMC also received speaker honoraria from PTC Therapeutics and BioMarin Pharmaceuticals and funding for contract research from Italpharmaco and Alpha Anomeric. RW, EK, CB, JvD and ND are (former) employees of BioMarin Nederland BV (formerly Prosensa Therapeutics BV) and performed the work with company budget in the form of salaries, equipment and facilities. JvD discloses being co-inventor on patents on exon skipping technology, some of which has been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents JvD is entitled to a share of royalties. JP, TB and TA are employees of Charles River Research Discovery Services in Finland and have no financial conflict of interest related to the submitted manuscript. Other authors have nothing to declare. The commercial affiliations of authors with Biomarin Pharmaceutical Ltd. and Charles River do not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
References
-
- Rader EP, Turk R, Willer T, Beltrán D, Inamori K-i, Peterson TA, et al. Role of dystroglycan in limiting contraction-induced injury to the sarcomeric cytoskeleton of mature skeletal muscle. 2016;113(39):10992–7. 10.1073/pnas.1605265113 %J Proceedings of the National Academy of Sciences. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
