

Griscelli Syndrome Type 2 Sine Albinism: Unraveling Differential RAB27A Effector Engagement
- PMID: 33362801
- PMCID: PMC7758216
- DOI: 10.3389/fimmu.2020.612977
Griscelli Syndrome Type 2 Sine Albinism: Unraveling Differential RAB27A Effector Engagement
Abstract
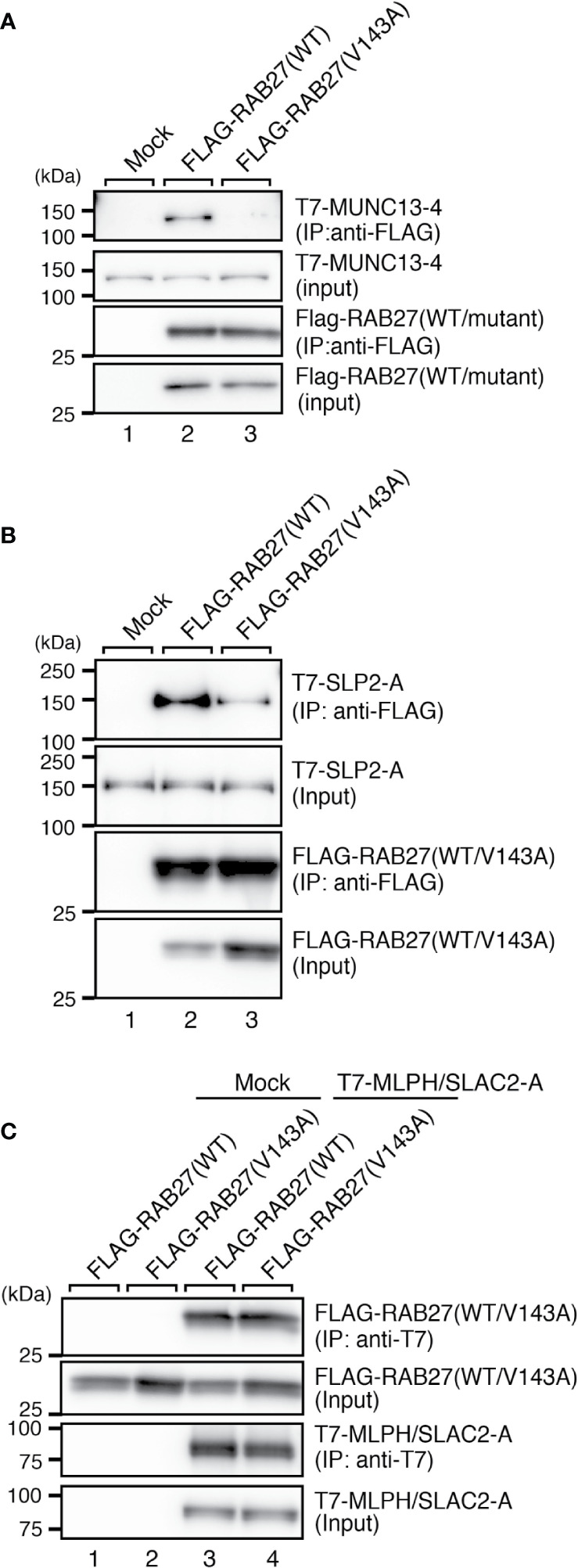
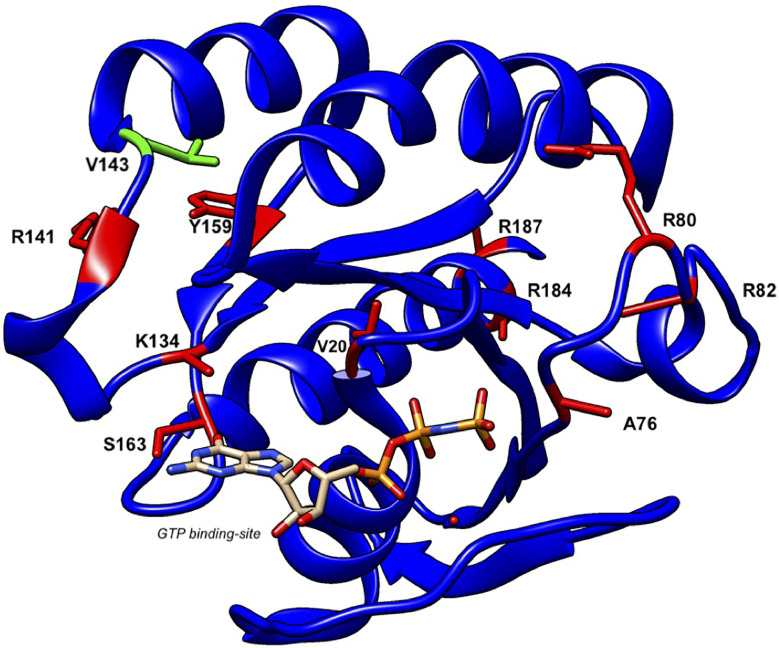
Griscelli syndrome type 2 (GS-2) is an inborn error of immunity characterized by partial albinism and episodes of hemophagocytic lymphohistiocytosis (HLH). It is caused by RAB27A mutations that encode RAB27A, a member of the Rab GTPase family. RAB27A is expressed in many tissues and regulates vesicular transport and organelle dynamics. Occasionally, GS-2 patients with RAB27A mutation display normal pigmentation. The study of such variants provides the opportunity to map distinct binding sites for tissue-specific effectors on RAB27A. Here we present a new case of GS-2 without albinism (GS-2 sine albinism) caused by a novel missense mutation (Val143Ala) in the RAB27A and characterize its functional cellular consequences. Using pertinent animal cell lines, the Val143Ala mutation impairs both the RAB27A-SLP2-A interaction and RAB27A-MUNC13-4 interaction, but it does not affect the RAB27A-melanophilin (MLPH)/SLAC2-A interaction that is crucial for skin and hair pigmentation. We conclude that disruption of the RAB27A-MUNC13-4 interaction in cytotoxic lymphocytes leads to the HLH predisposition of the GS-2 patient with the Val143Ala mutation. Finally, we include a review of GS-2 sine albinism cases reported in the literature, summarizing their genetic and clinical characteristics.
Keywords: Griscelli syndrome type 2 sine albinism; MLPH/SLAC2-A; MUNC13-4; RAB27A; hemophagocytic lymphohistiocytosis; inborn error of immunity; whole-exome sequencing.
Copyright © 2020 Ohishi, Ammann, Ziaee, Strege, Groß, Amos, Shahrooei, Ashournia, Razaghian, Griffiths, Ehl, Fukuda and Parvaneh.
Conflict of interest statement
SA worked as a scientific advisor for Sobi. The IRB of Children’s Medical Center affiliated to TUMS approved this study (IR.TUMS.CHMC.REC.1399.080). The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer HK declared a past co-authorship with one of the authors SE to the handling editor. The reviewer AF declared a past co-authorship with one of the authors SE to the handling editor.
Figures
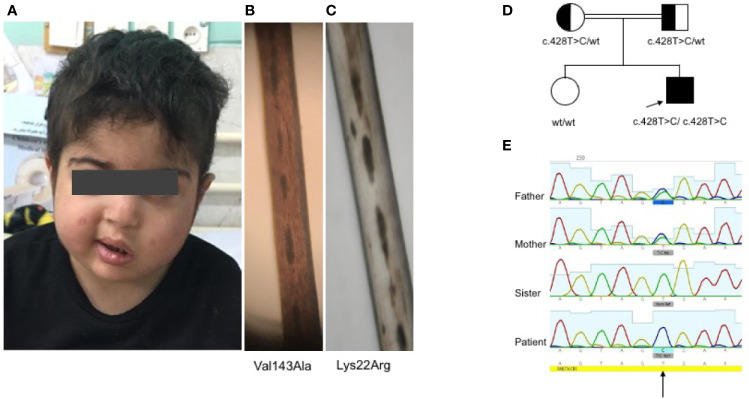
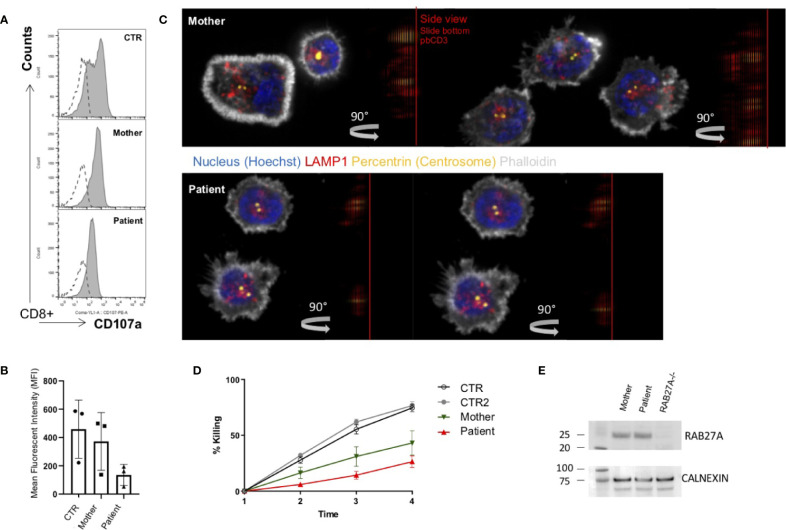
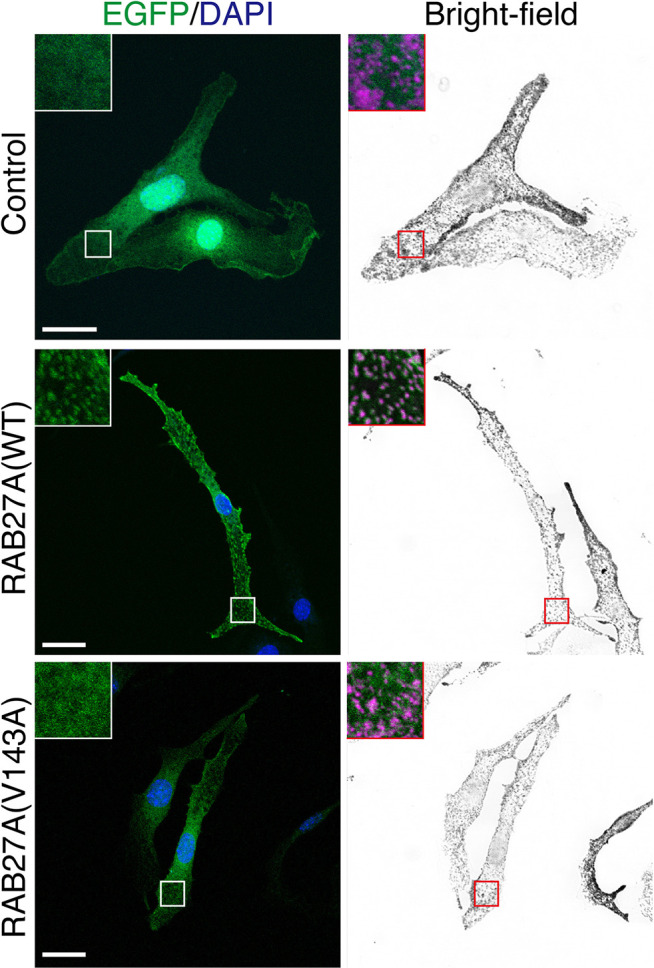
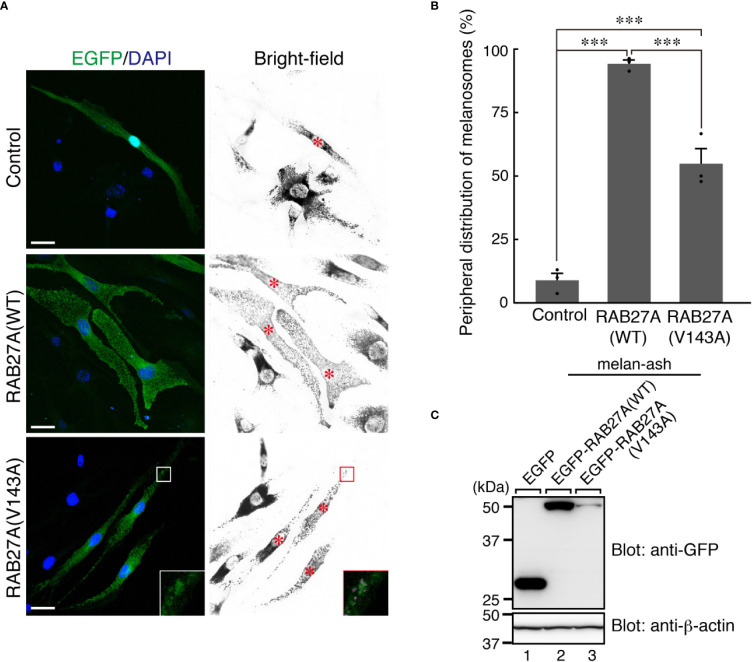
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous