

Epigenetic Variation Analysis Leads to Biomarker Discovery in Gastric Adenocarcinoma
- PMID: 33363566
- PMCID: PMC7753064
- DOI: 10.3389/fgene.2020.551787
Epigenetic Variation Analysis Leads to Biomarker Discovery in Gastric Adenocarcinoma
Abstract
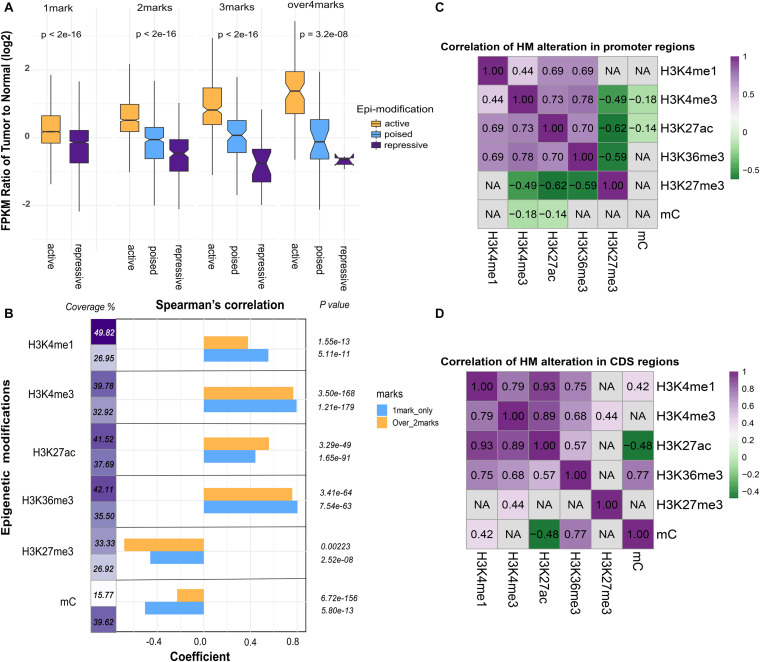
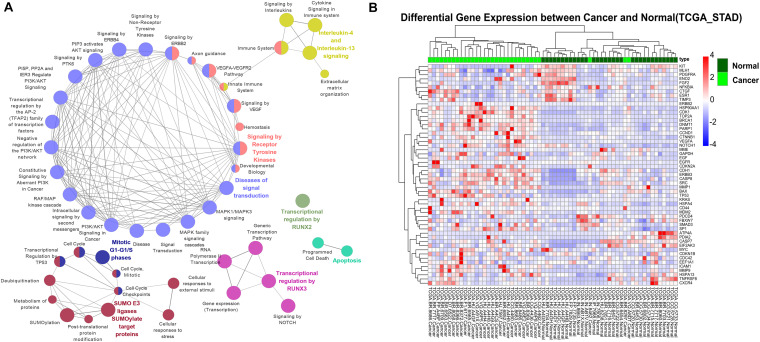
As one of the most common malignant tumors worldwide, gastric adenocarcinoma (GC) and its prognosis are still poorly understood. Various genetic and epigenetic factors have been indicated in GC carcinogenesis. However, a comprehensive and in-depth investigation of epigenetic alteration in gastric cancer is still missing. In this study, we systematically investigated some key epigenetic features in GC, including DNA methylation and five core histone modifications. Data from The Cancer Genome Atlas Program and other studies (Gene Expression Omnibus) were collected, analyzed, and validated with multivariate statistical analysis methods. The landscape of epi-modifications in gastric cancer was described. Chromatin state transition analysis showed a histone marker shift in gastric cancer genome by employing a Hidden-Markov-Model based approach, indicated that histone marks tend to label different sets of genes in GC compared to control. An additive effect of these epigenetic marks was observed by integrated analysis with gene expression data, suggesting epigenetic modifications may cooperatively regulate gene expression. However, the effect of DNA methylation was found more significant without the presence of the five histone modifications in our study. By constructing a PPI network, key genes to distinguish GC from normal samples were identified, and distinct patterns of oncogenic pathways in GC were revealed. Some of these genes can also serve as potential biomarkers to classify various GC molecular subtypes. Our results provide important insights into the epigenetic regulation in gastric cancer and other cancers in general. This study describes the aberrant epigenetic variation pattern in GC and provides potential direction for epigenetic biomarker discovery.
Keywords: DNA methylation; biomarker; epigenetics; gastric cancer; histone modification.
Copyright © 2020 Zhang and Guo.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous