

Identifying epitopes for cluster of differentiation and design of new peptides inhibitors against human SARS-CoV-2 spike RBD by an in-silico approach
- PMID: 33364503
- PMCID: PMC7753134
- DOI: 10.1016/j.heliyon.2020.e05739
Identifying epitopes for cluster of differentiation and design of new peptides inhibitors against human SARS-CoV-2 spike RBD by an in-silico approach
Abstract
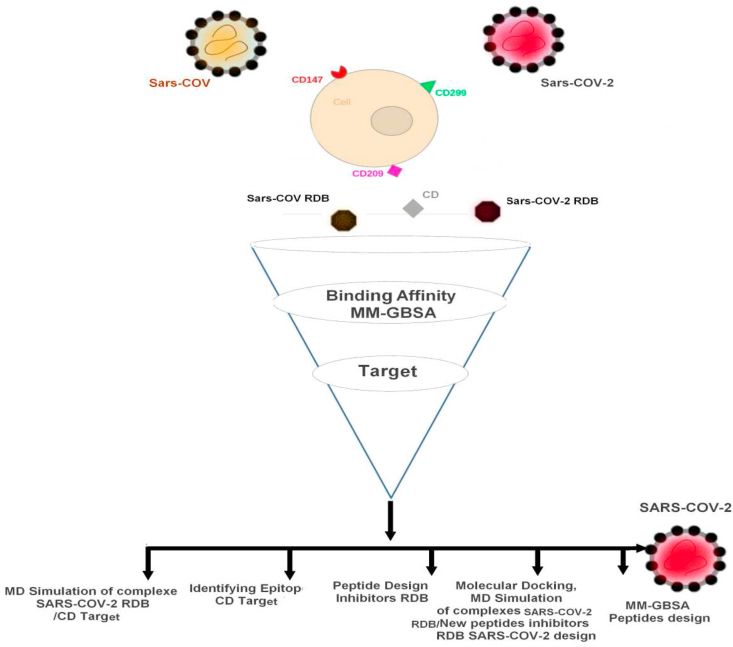
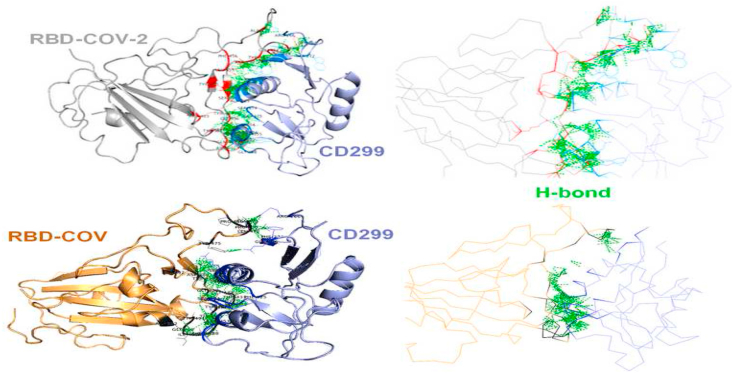
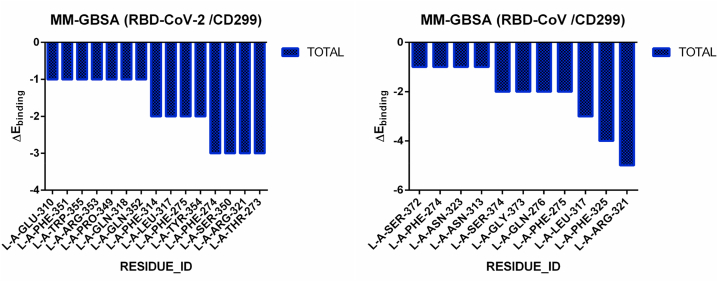
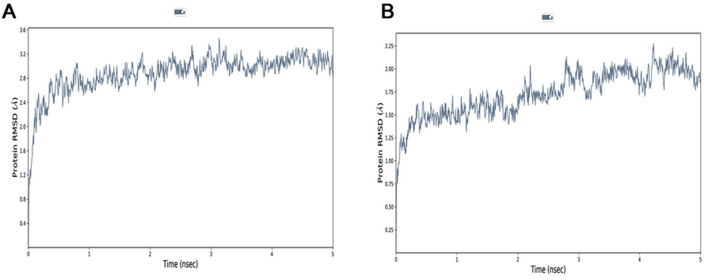
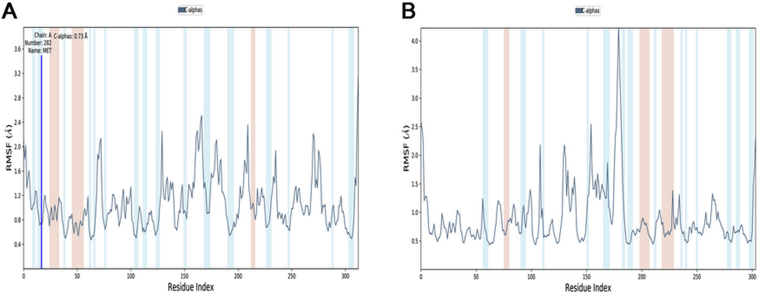
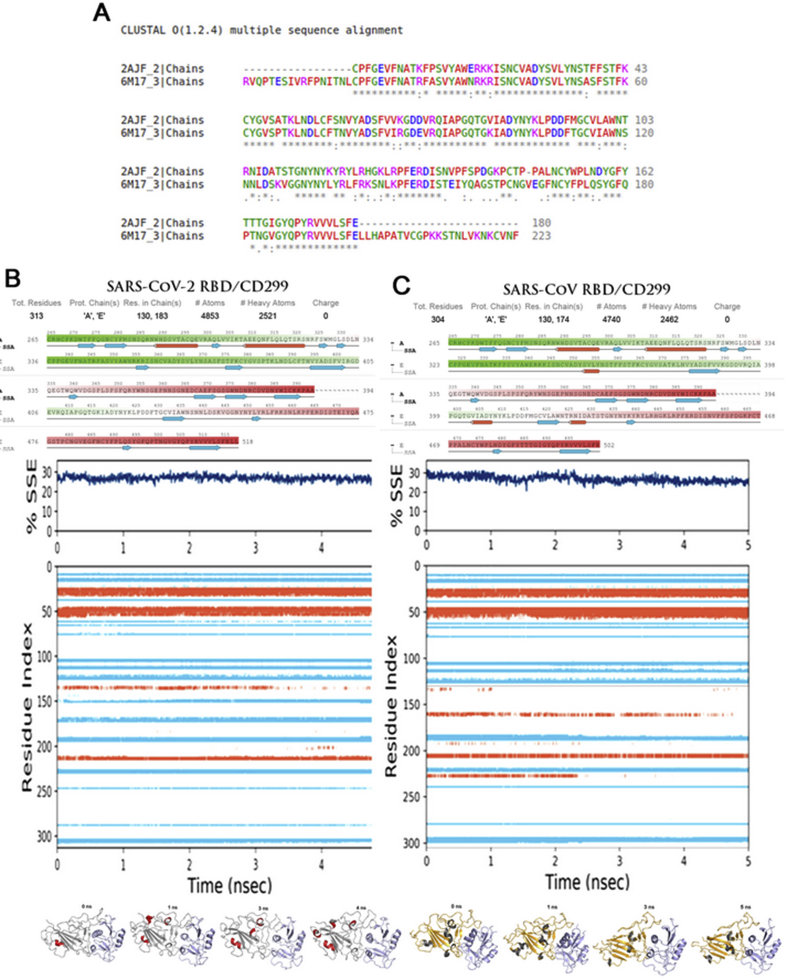
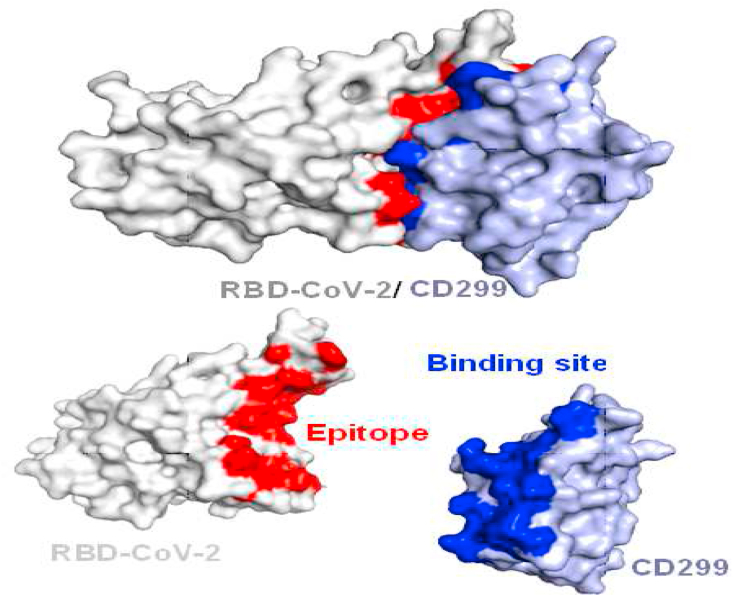
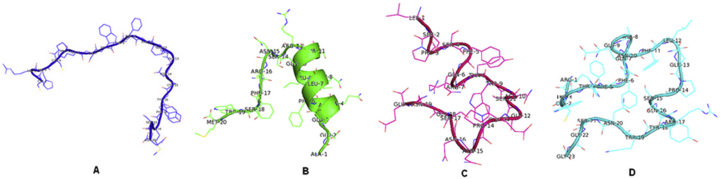
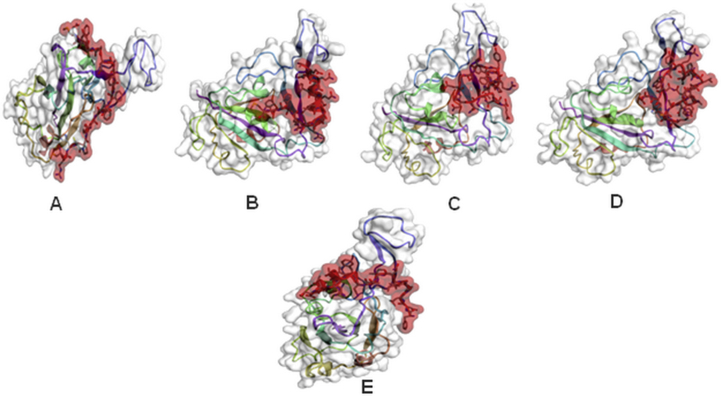
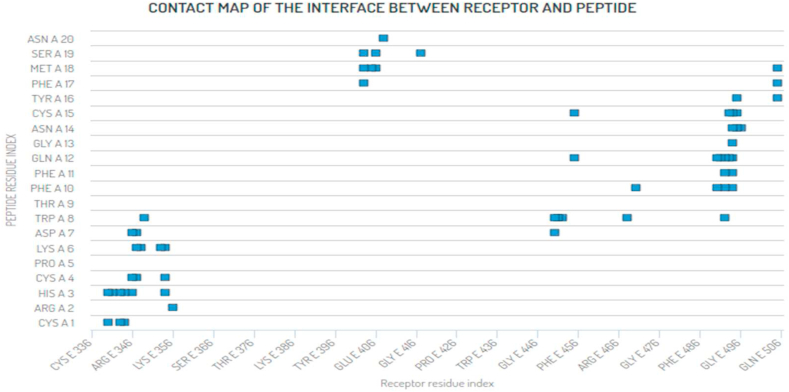
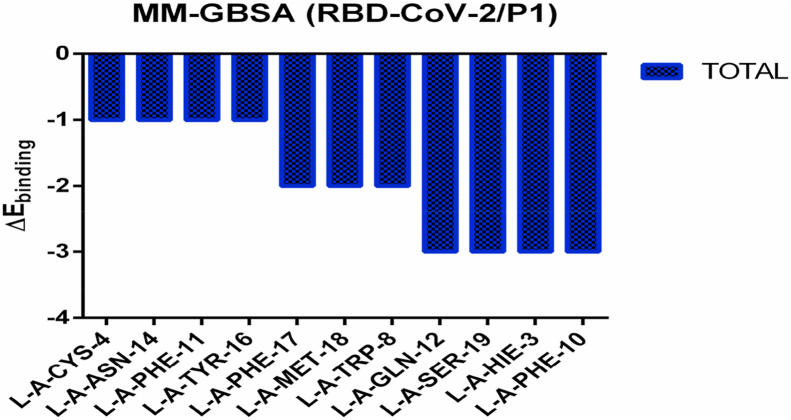
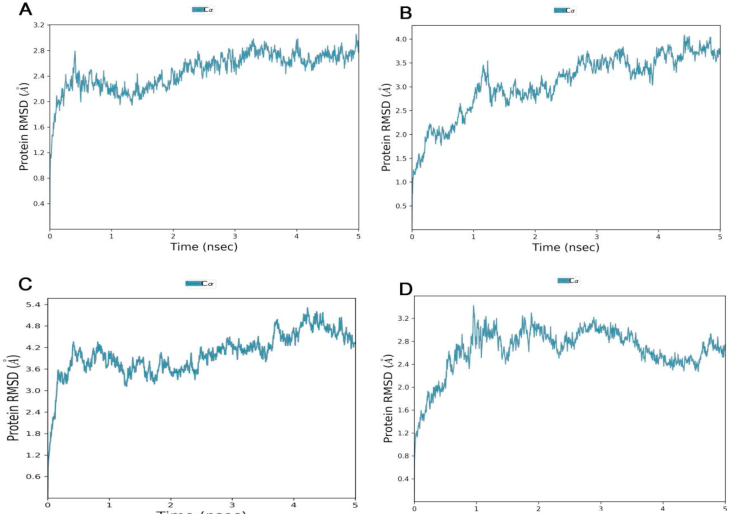
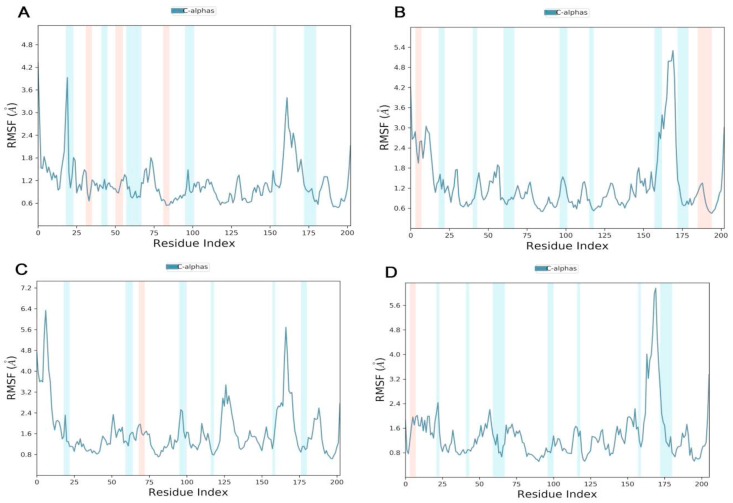
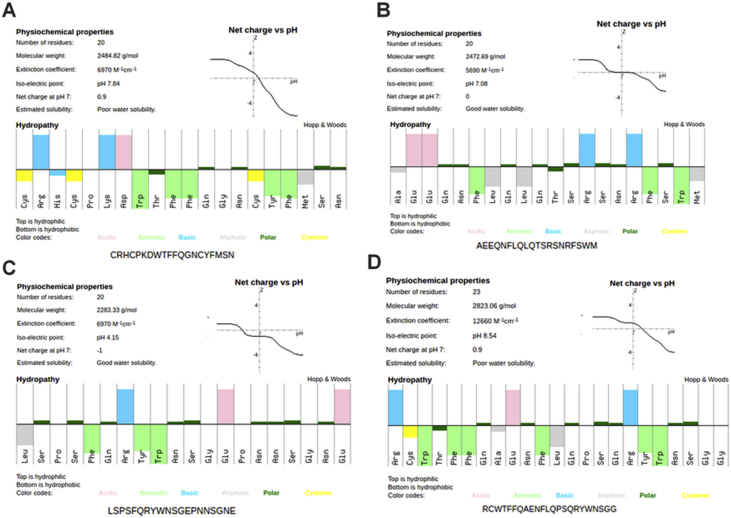
The coronavirus disease 19 (COVID-19) is a highly contagious and rapidly spreading infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). In some cases, the disease can be fatal which resulted in more than one million deaths worldwide according the WHO. Currently, there is no effective vaccine or treatment for COVID-19, however many small-molecule inhibitors have shown potent antiviral activity against SARS-CoV-2 and some of them are now under clinical trials. Despite their promising activities, the development of these small molecules for the clinical use can be limited by many factors like the off-target effect, the poor stability, and the low bioavailability. The clusters of differentiation CD147, CD209, CD299 have been identified as essential entry co-receptors for SARS-CoV-2 species specificity to humans, although the underlying mechanisms are yet to be fully elucidated. In this paper, protein-protein docking was utilized for identifying the critical epitopes in CD147, CD209 and CD299 which are involved in the binding with SARS-CoV-2 Spike receptor binding domain (RBD). The results of binding free energies showed a high affinity of SARS-CoV-2 RBD to CD299 receptor which was used as a reference to derive hypothetical peptide sequences with specific binding activities to SARS-CoV-2 RBD. Molecular docking and molecular dynamics simulations of the newly designed peptides showed favorable binding features and stability with SARS-CoV-2 RBD and therefore can be further considered as potential candidates in future anti-SARS CoV-2 drug discovery studies.
Keywords: Biological sciences; Chemistry; Cluster of differentiation; Computer science; Coronavirus 19; Engineering; Molecular docking; Molecular dynamics; Peptide-based drugs; Physics; Spike protein.
© 2020 Published by Elsevier Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
A Novel Therapeutic Peptide Blocks SARS-CoV-2 Spike Protein Binding with Host Cell ACE2 Receptor.Drugs R D. 2021 Sep;21(3):273-283. doi: 10.1007/s40268-021-00357-0. Epub 2021 Jul 29. Drugs R D. 2021. PMID: 34324175 Free PMC article.
-
Structural Basis of a Human Neutralizing Antibody Specific to the SARS-CoV-2 Spike Protein Receptor-Binding Domain.Microbiol Spectr. 2021 Oct 31;9(2):e0135221. doi: 10.1128/Spectrum.01352-21. Epub 2021 Oct 13. Microbiol Spectr. 2021. PMID: 34643438 Free PMC article.
-
Screening of inhibitors against SARS-CoV-2 spike protein and their capability to block the viral entry mechanism: A viroinformatics study.Saudi J Biol Sci. 2021 Jun;28(6):3262-3269. doi: 10.1016/j.sjbs.2021.02.066. Epub 2021 Feb 26. Saudi J Biol Sci. 2021. PMID: 33654454 Free PMC article.
-
Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses.Drug Resist Updat. 2020 Dec;53:100721. doi: 10.1016/j.drup.2020.100721. Epub 2020 Aug 26. Drug Resist Updat. 2020. PMID: 33132205 Free PMC article. Review.
-
Therapeutic potential of green tea catechin, (-)-epigallocatechin-3-O-gallate (EGCG) in SARS-CoV-2 infection: Major interactions with host/virus proteases.Phytomed Plus. 2023 Feb;3(1):100402. doi: 10.1016/j.phyplu.2022.100402. Epub 2022 Dec 30. Phytomed Plus. 2023. PMID: 36597465 Free PMC article. Review.
Cited by
-
Designing of a bispecific antibody against SARS-CoV-2 spike glycoprotein targeting human entry receptors DPP4 and ACE2.Hum Immunol. 2022 Apr;83(4):346-355. doi: 10.1016/j.humimm.2022.01.004. Epub 2022 Jan 10. Hum Immunol. 2022. PMID: 35042653 Free PMC article.
-
Myeloid-Derived Suppressor Cells (MDSCs) and Obesity-Induced Inflammation in Type 2 Diabetes.Diagnostics (Basel). 2024 Nov 1;14(21):2453. doi: 10.3390/diagnostics14212453. Diagnostics (Basel). 2024. PMID: 39518420 Free PMC article. Review.
-
Comparative genomics, evolutionary epidemiology, and RBD-hACE2 receptor binding pattern in B.1.1.7 (Alpha) and B.1.617.2 (Delta) related to their pandemic response in UK and India.Infect Genet Evol. 2022 Jul;101:105282. doi: 10.1016/j.meegid.2022.105282. Epub 2022 Apr 13. Infect Genet Evol. 2022. PMID: 35427787 Free PMC article.
-
Molecular modeling of the interaction of ligands with ACE2-SARS-CoV-2 spike protein complex.In Silico Pharmacol. 2021 Oct 7;9(1):55. doi: 10.1007/s40203-021-00114-w. eCollection 2021. In Silico Pharmacol. 2021. PMID: 34631362 Free PMC article.
-
Protein interactions with metallothionein-3 promote vectorial active transport in human proximal tubular cells.PLoS One. 2022 May 3;17(5):e0267599. doi: 10.1371/journal.pone.0267599. eCollection 2022. PLoS One. 2022. PMID: 35503771 Free PMC article.
References
-
- Munster V.J., Koopmans M., van Doremalen N., van Riel D., de Wit E. A novel coronavirus emerging in China—key questions for impact assessment. N. Engl. J. Med. 2020;382(8):692–694. - PubMed
-
- Shiva Raeisi D, Seyed Ali HZ, Zahra G, Sepideh A. 2020. Covid-19 (SARS-CoV-2) VS Sars-CoV; Summary of all things that healthcare providers should know. Am. J. Biomed. Sci. Res. 2020 ; 376-382. .
LinkOut - more resources
Full Text Sources
Miscellaneous
