

Variation Patterns of NLR Clusters in Arabidopsis thaliana Genomes
- PMID: 33367252
- PMCID: PMC7747988
- DOI: 10.1016/j.xplc.2020.100089
Variation Patterns of NLR Clusters in Arabidopsis thaliana Genomes
Abstract
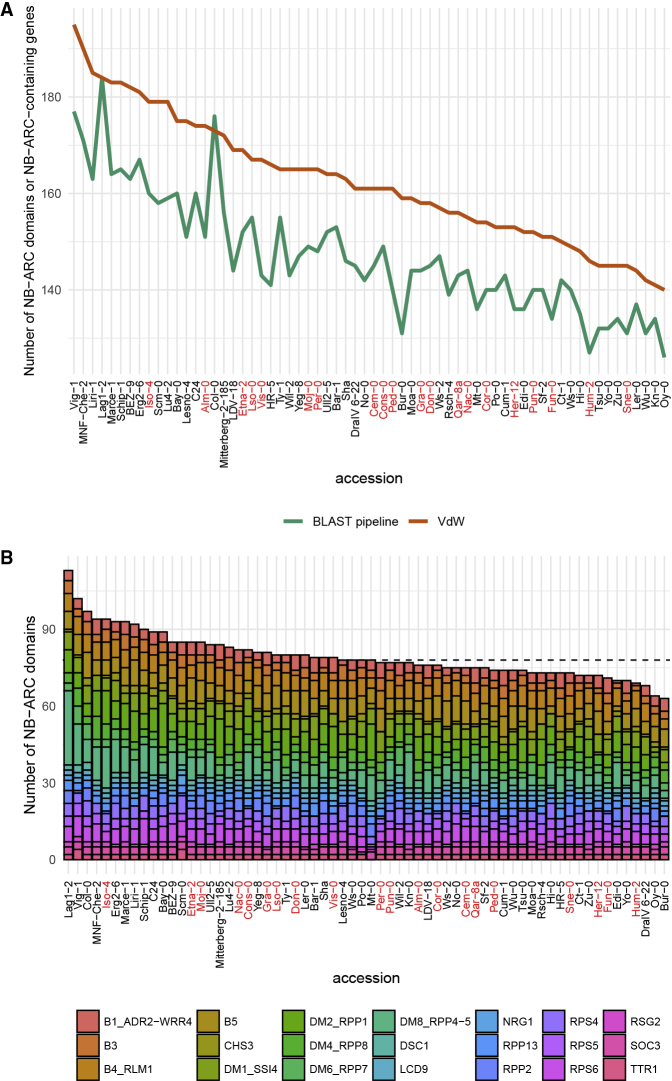
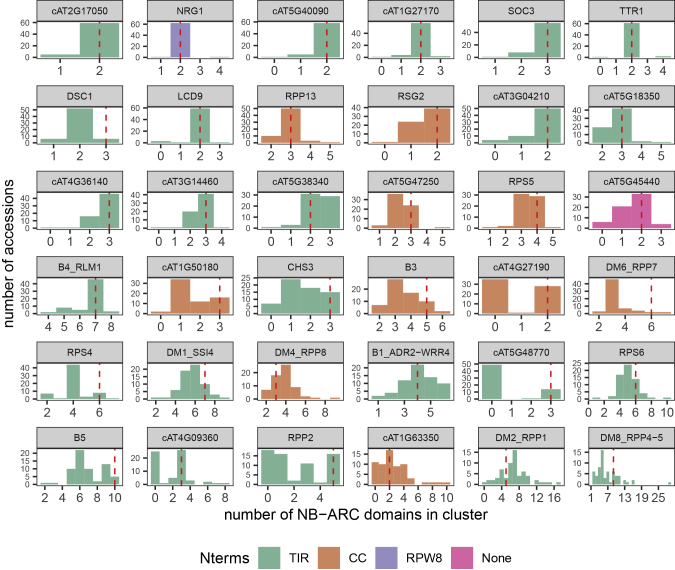
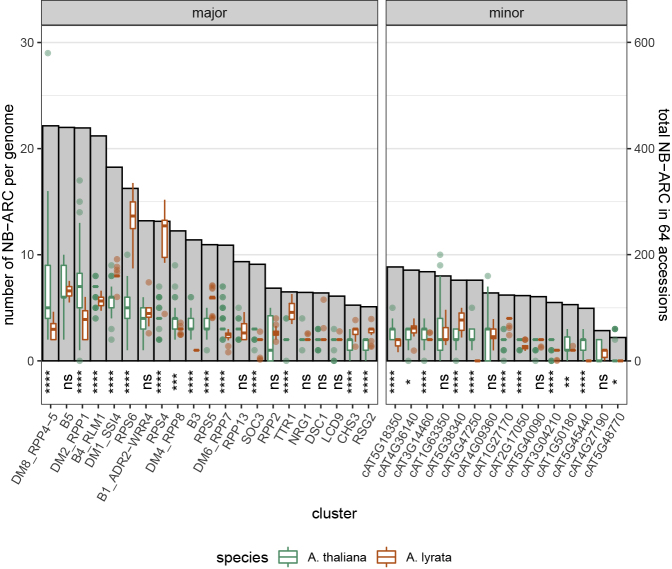
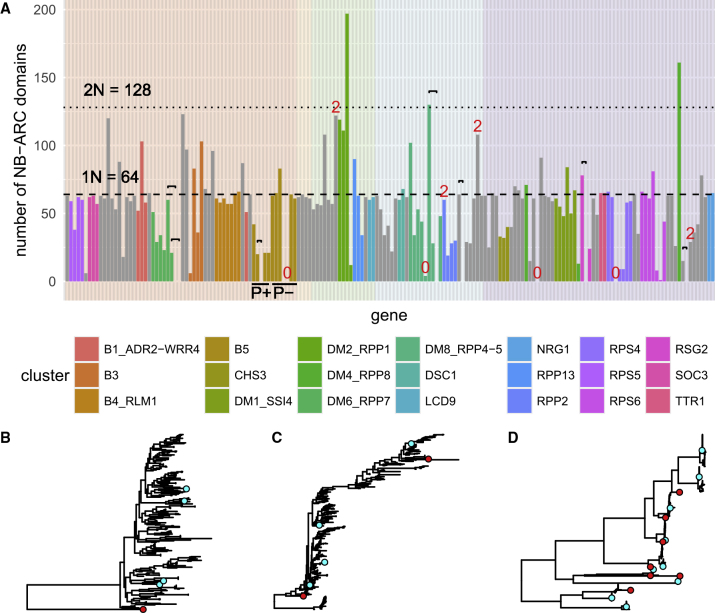
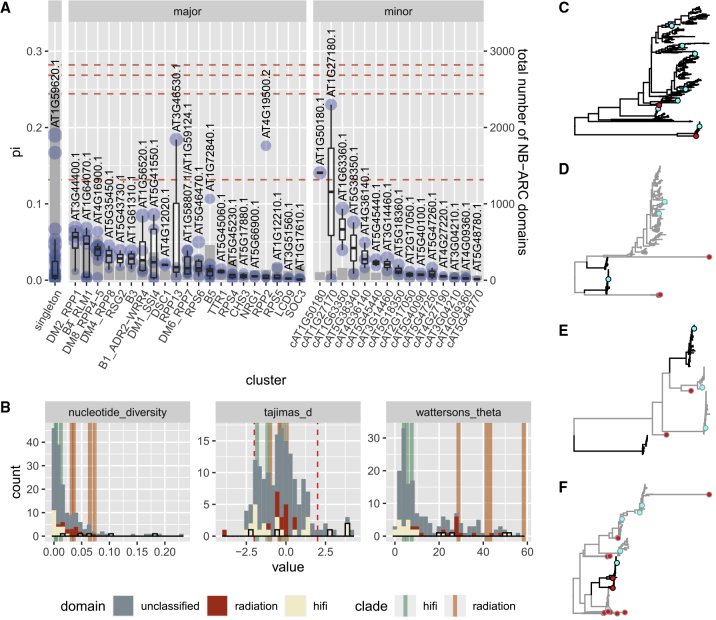
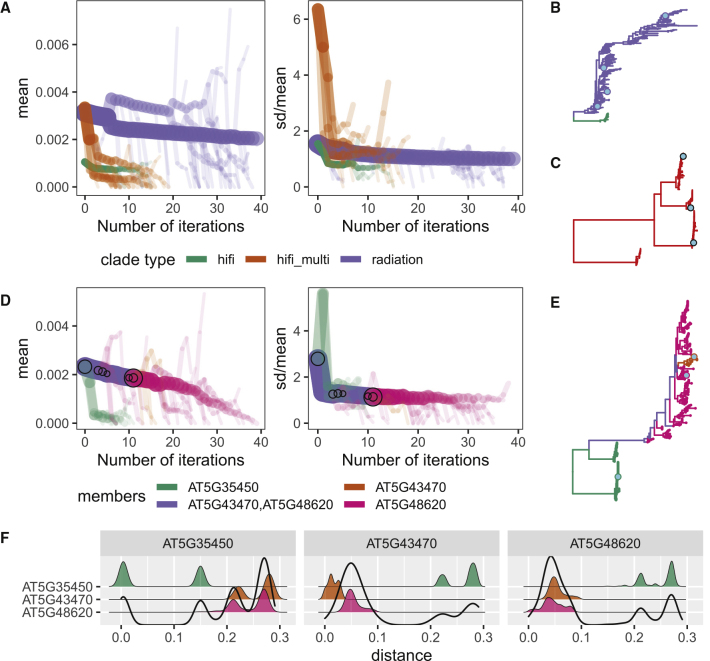
The nucleotide-binding domain and leucine-rich repeat (NLR) gene family is highly expanded in the plant lineage with extensive sequence and structure polymorphisms. To survey the landscape of NLR expansion, we mined the published long-read data generated by the resistance gene enrichment sequencing of 64 diverse Arabidopsis thaliana accessions. We found that the hot spots of massive multi-gene NLR cluster expansion did not typically span the whole cluster; instead, they were restricted to a handful of, or only one, dominant radiation(s). All sequences in such a radiation were distinct from other genes in the cluster but not from each other in the clade, making it difficult to assign trustworthy reference-based orthologies when multiple reference genes were present in the radiation. Consequently, NLR genes can be broadly divided into two types: radiating or high-fidelity, where high-fidelity genes are well conserved and well separated from other clades. A similar distinction could be made for NLR clusters, depending on whether cluster size was determined primarily by extensive radiation or the presence of numerous high-fidelity genes. We also identified groups of well-conserved NLR clades that were missing from the Columbia-0 reference genome. This suggests that the classification of NLRs using gene IDs from a single reference accession can rarely capture all major paralogs in a cluster accurately and representatively and that a reference-agnostic perspective is required to properly characterize these additional variations. Finally, we present a quantitative visualization method for differentiating these situations in a given clade of interest.
Keywords: NLR; cluster; disease resistance; evolution; phylogenetics; plant immunity.
© 2020 The Author(s).
Figures
References
-
- Adachi H., Derevnina L., Kamoun S. NLR singletons, pairs, and networks: evolution, assembly, and regulation of the intracellular immunoreceptor circuitry of plants. Curr. Opin. Plant Biol. 2019;50:121–131. - PubMed
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- van der Biezen E.A., Jones J.D. The NB-ARC domain: a novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr. Biol. 1998;8:R226–R227. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
