

Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B
- PMID: 33370413
- PMCID: PMC7853461
- DOI: 10.1371/journal.ppat.1009181
Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B
Abstract
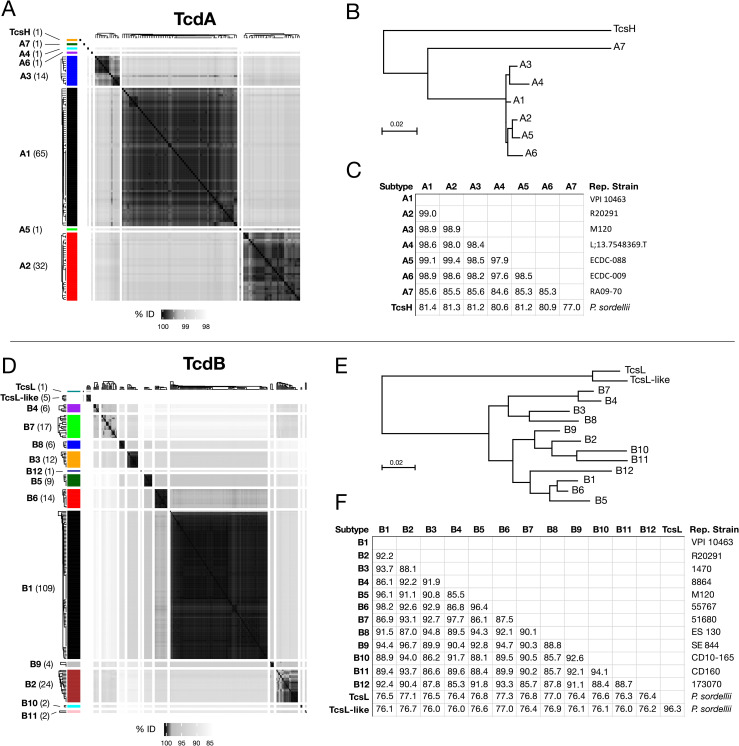
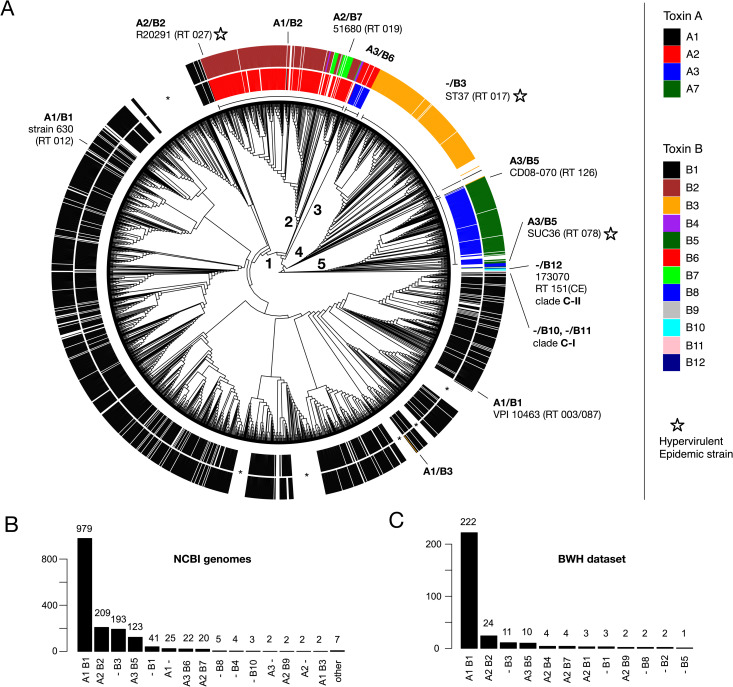
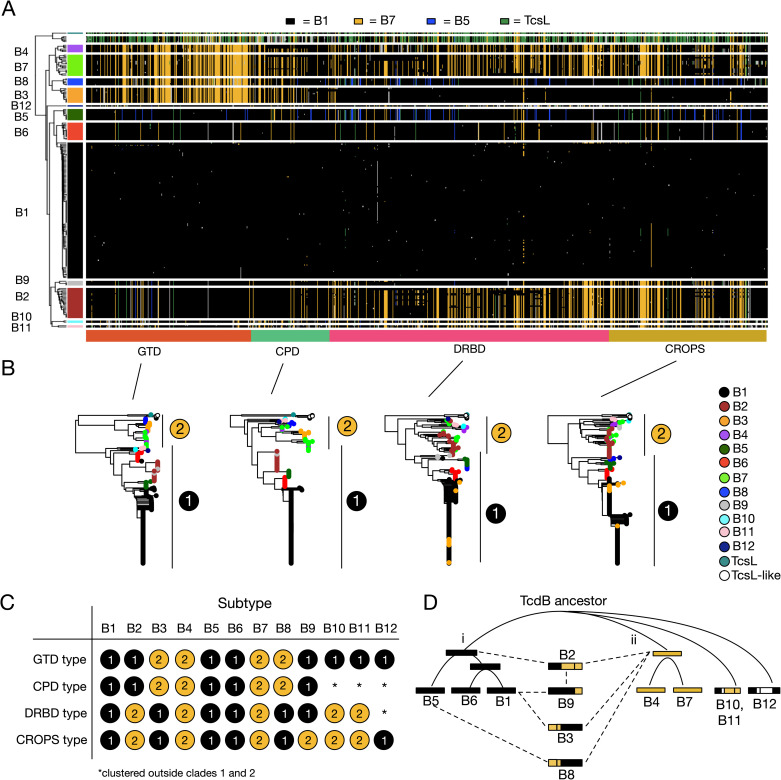
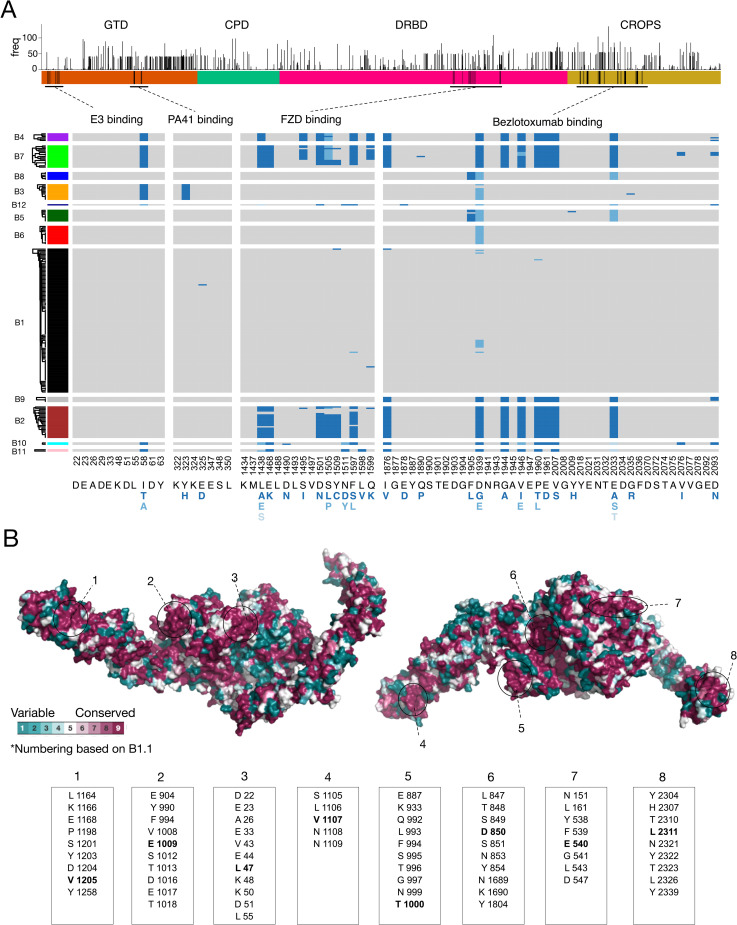
Clostridioides difficile is the major worldwide cause of antibiotic-associated gastrointestinal infection. A pathogenicity locus (PaLoc) encoding one or two homologous toxins, toxin A (TcdA) and toxin B (TcdB), is essential for C. difficile pathogenicity. However, toxin sequence variation poses major challenges for the development of diagnostic assays, therapeutics, and vaccines. Here, we present a comprehensive phylogenomic analysis of 8,839 C. difficile strains and their toxins including 6,492 genomes that we assembled from the NCBI short read archive. A total of 5,175 tcdA and 8,022 tcdB genes clustered into 7 (A1-A7) and 12 (B1-B12) distinct subtypes, which form the basis of a new method for toxin-based subtyping of C. difficile. We developed a haplotype coloring algorithm to visualize amino acid variation across all toxin sequences, which revealed that TcdB has diversified through extensive homologous recombination throughout its entire sequence, and formed new subtypes through distinct recombination events. In contrast, TcdA varies mainly in the number of repeats in its C-terminal repetitive region, suggesting that recombination-mediated diversification of TcdB provides a selective advantage in C. difficile evolution. The application of toxin subtyping is then validated by classifying 351 C. difficile clinical isolates from Brigham and Women's Hospital in Boston, demonstrating its clinical utility. Subtyping partitions TcdB into binary functional and antigenic groups generated by intragenic recombinations, including two distinct cell-rounding phenotypes, whether recognizing frizzled proteins as receptors, and whether it can be efficiently neutralized by monoclonal antibody bezlotoxumab, the only FDA-approved therapeutic antibody. Our analysis also identifies eight universally conserved surface patches across the TcdB structure, representing ideal targets for developing broad-spectrum therapeutics. Finally, we established an open online database (DiffBase) as a central hub for collection and classification of C. difficile toxins, which will help clinicians decide on therapeutic strategies targeting specific toxin variants, and allow researchers to monitor the ongoing evolution and diversification of C. difficile.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Genomic and phenotypic studies among Clostridioides difficile isolates show a high prevalence of clade 2 and great diversity in clinical isolates from Mexican adults and children with healthcare-associated diarrhea.Microbiol Spectr. 2024 Jul 2;12(7):e0394723. doi: 10.1128/spectrum.03947-23. Epub 2024 Jun 12. Microbiol Spectr. 2024. PMID: 38864670 Free PMC article.
-
Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B.Commun Biol. 2020 Jul 3;3(1):347. doi: 10.1038/s42003-020-1078-y. Commun Biol. 2020. PMID: 32620855 Free PMC article.
-
Comparative genomics of Clostridioides difficile toxinotypes identifies module-based toxin gene evolution.Microb Genom. 2020 Oct;6(10):mgen000449. doi: 10.1099/mgen.0.000449. Microb Genom. 2020. PMID: 33030421 Free PMC article.
-
The Importance of Therapeutically Targeting the Binary Toxin from Clostridioides difficile.Int J Mol Sci. 2021 Mar 13;22(6):2926. doi: 10.3390/ijms22062926. Int J Mol Sci. 2021. PMID: 33805767 Free PMC article. Review.
-
Clostridioides difficile Toxins: Host Cell Interactions and Their Role in Disease Pathogenesis.Toxins (Basel). 2024 May 24;16(6):241. doi: 10.3390/toxins16060241. Toxins (Basel). 2024. PMID: 38922136 Free PMC article. Review.
Cited by
-
Bacterial toxins induce non-canonical migracytosis to aggravate acute inflammation.Cell Discov. 2024 Nov 5;10(1):112. doi: 10.1038/s41421-024-00729-1. Cell Discov. 2024. PMID: 39500876 Free PMC article.
-
A multivalent mRNA-LNP vaccine protects against Clostridioides difficile infection.Science. 2024 Oct 4;386(6717):69-75. doi: 10.1126/science.adn4955. Epub 2024 Oct 3. Science. 2024. PMID: 39361752 Free PMC article.
-
Genomic and phenotypic studies among Clostridioides difficile isolates show a high prevalence of clade 2 and great diversity in clinical isolates from Mexican adults and children with healthcare-associated diarrhea.Microbiol Spectr. 2024 Jul 2;12(7):e0394723. doi: 10.1128/spectrum.03947-23. Epub 2024 Jun 12. Microbiol Spectr. 2024. PMID: 38864670 Free PMC article.
-
Commensal-pathogen dynamics structure disease outcomes during Clostridioides difficile colonization.Cell Host Microbe. 2025 Jan 8;33(1):30-41.e6. doi: 10.1016/j.chom.2024.12.002. Epub 2024 Dec 27. Cell Host Microbe. 2025. PMID: 39731916
-
Against Clostridioides difficile Infection: An Update on Vaccine Development.Toxins (Basel). 2025 May 1;17(5):222. doi: 10.3390/toxins17050222. Toxins (Basel). 2025. PMID: 40423305 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
