

Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants
- PMID: 33371482
- PMCID: PMC7767499
- DOI: 10.3390/biom10121702
Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants
Abstract
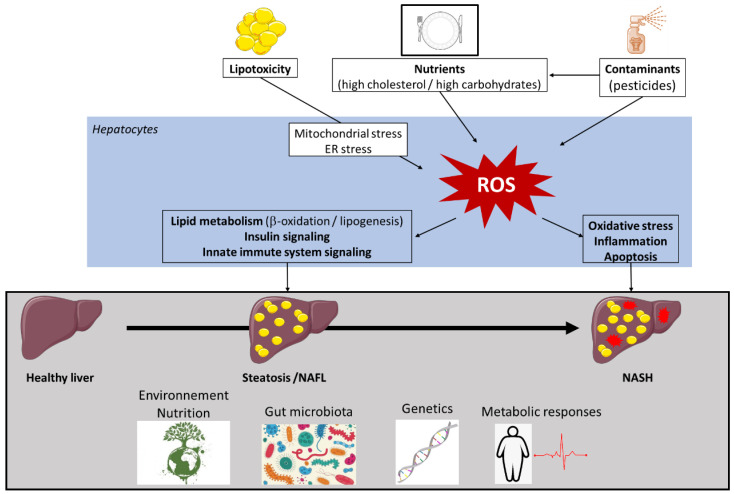
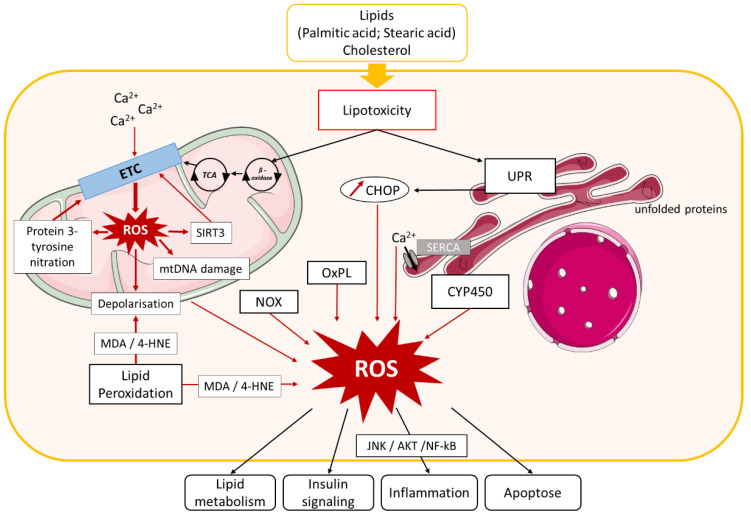
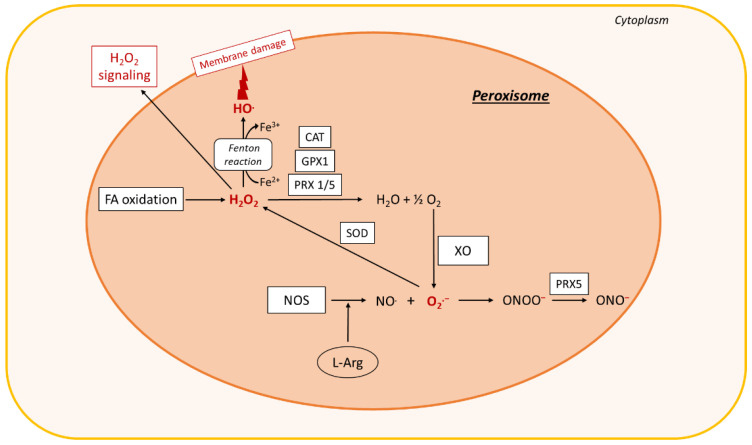
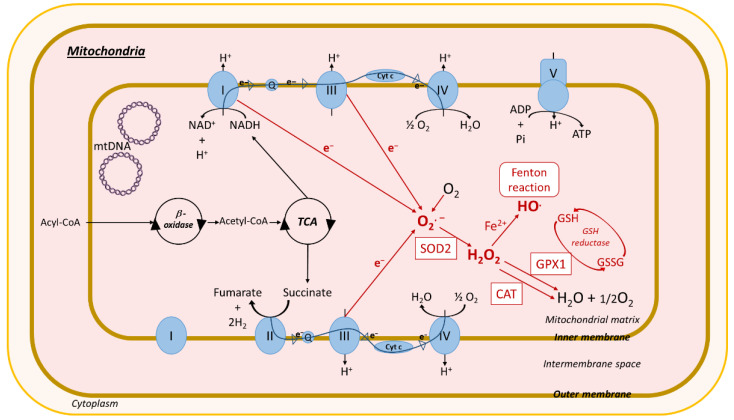
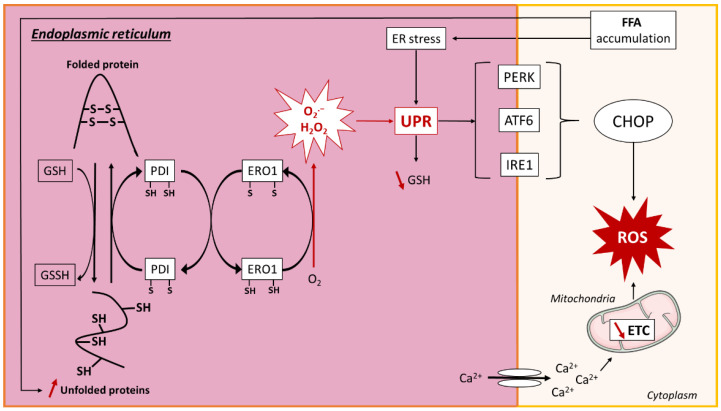
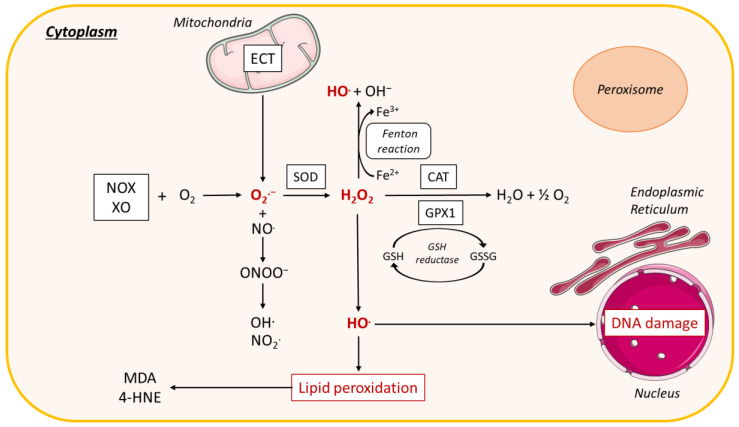
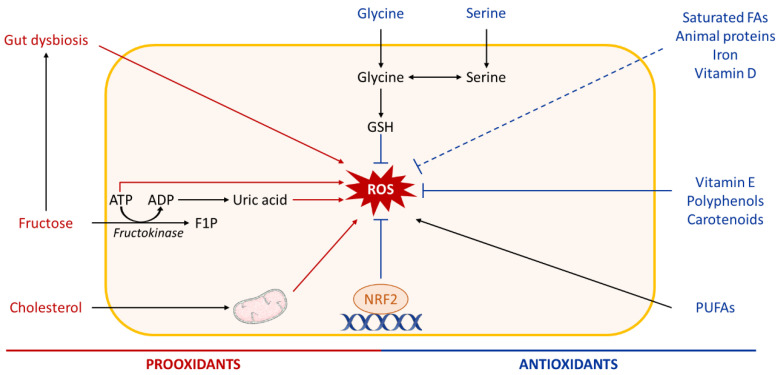
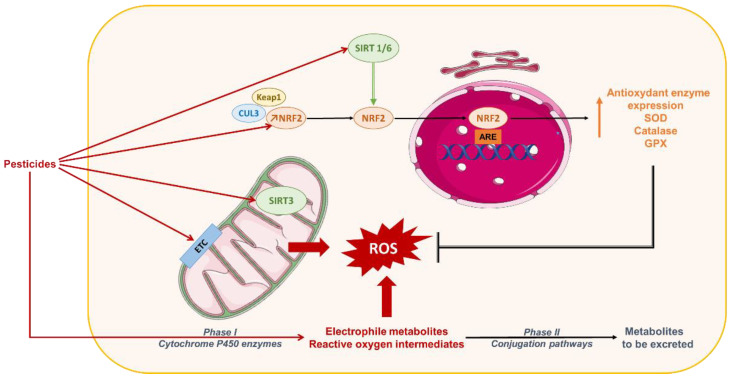
Non-alcoholic fatty liver disease (NAFLD) is often the hepatic expression of metabolic syndrome and its comorbidities that comprise, among others, obesity and insulin-resistance. NAFLD involves a large spectrum of clinical conditions. These range from steatosis, a benign liver disorder characterized by the accumulation of fat in hepatocytes, to non-alcoholic steatohepatitis (NASH), which is characterized by inflammation, hepatocyte damage, and liver fibrosis. NASH can further progress to cirrhosis and hepatocellular carcinoma. The etiology of NAFLD involves both genetic and environmental factors, including an unhealthy lifestyle. Of note, unhealthy eating is clearly associated with NAFLD development and progression to NASH. Both macronutrients (sugars, lipids, proteins) and micronutrients (vitamins, phytoingredients, antioxidants) affect NAFLD pathogenesis. Furthermore, some evidence indicates disruption of metabolic homeostasis by food contaminants, some of which are risk factor candidates in NAFLD. At the molecular level, several models have been proposed for the pathogenesis of NAFLD. Most importantly, oxidative stress and mitochondrial damage have been reported to be causative in NAFLD initiation and progression. The aim of this review is to provide an overview of the contribution of nutrients and food contaminants, especially pesticides, to oxidative stress and how they may influence NAFLD pathogenesis.
Keywords: food contaminant; macronutrients; micronutrients; mitochondria; non-alcoholic fatty liver disease (NAFLD); non-alcoholic steatohepatitis (NASH); oxidative stress; reactive oxygen species (ROS); steatosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
