

AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?
- PMID: 33375416
- PMCID: PMC7795930
- DOI: 10.3390/ijms22010186
AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?
Abstract
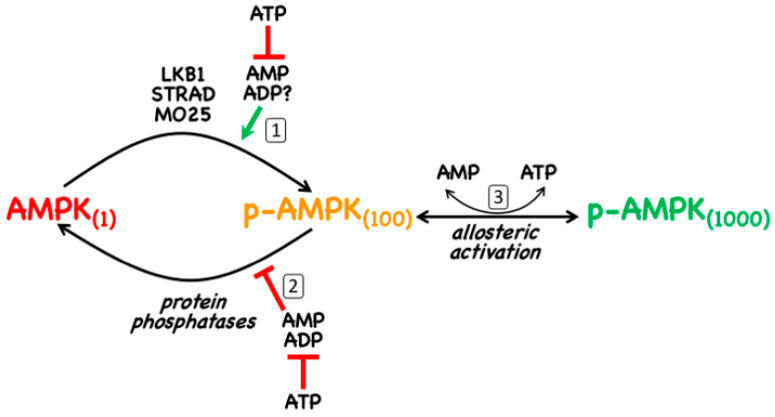
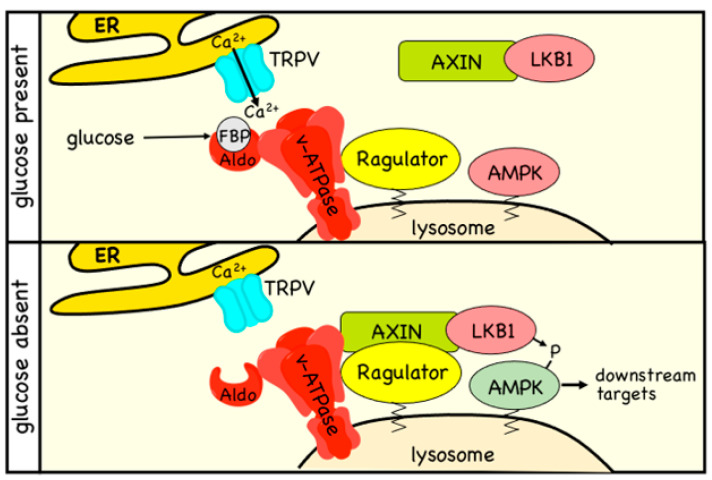
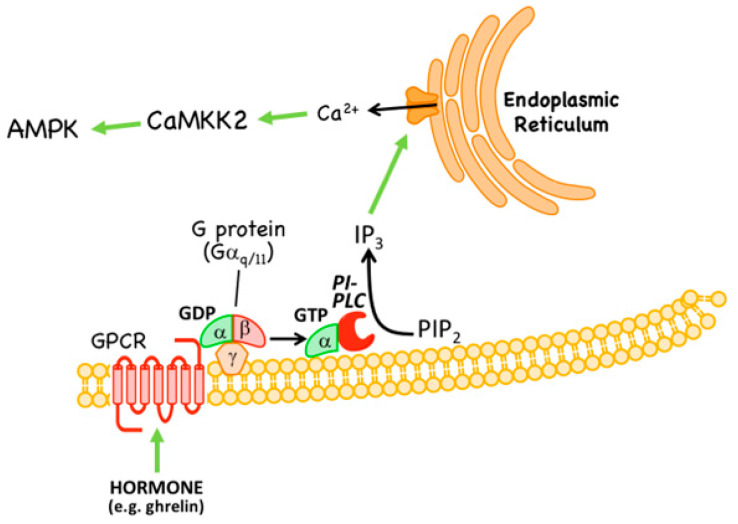
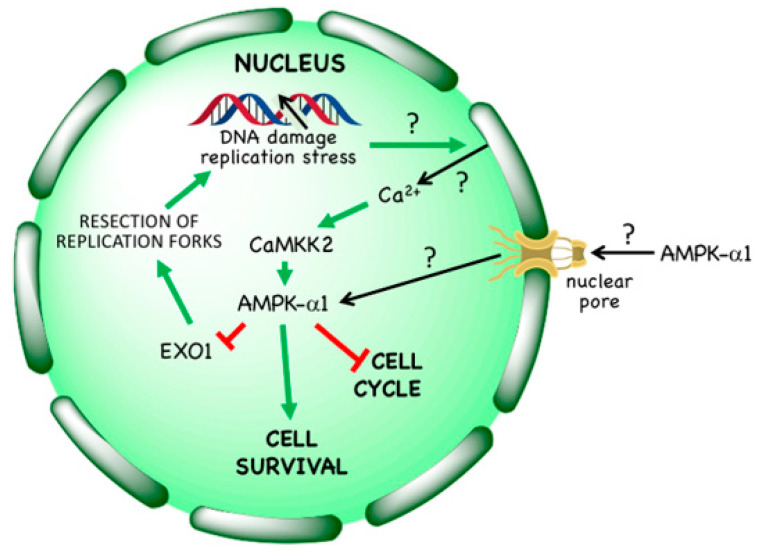
AMP-activated protein kinase (AMPK) is a key regulator of cellular energy balance. In response to metabolic stress, it acts to redress energy imbalance through promotion of ATP-generating catabolic processes and inhibition of ATP-consuming processes, including cell growth and proliferation. While findings that AMPK was a downstream effector of the tumour suppressor LKB1 indicated that it might act to repress tumourigenesis, more recent evidence suggests that AMPK can either suppress or promote cancer, depending on the context. Prior to tumourigenesis AMPK may indeed restrain aberrant growth, but once a cancer has arisen, AMPK may instead support survival of the cancer cells by adjusting their rate of growth to match their energy supply, as well as promoting genome stability. The two isoforms of the AMPK catalytic subunit may have distinct functions in human cancers, with the AMPK-α1 gene often being amplified, while the AMPK-α2 gene is more often mutated. The prevalence of metabolic disorders, such as obesity and Type 2 diabetes, has led to the development of a wide range of AMPK-activating drugs. While these might be useful as preventative therapeutics in individuals predisposed to cancer, it seems more likely that AMPK inhibitors, whose development has lagged behind that of activators, would be efficacious for the treatment of pre-existing cancers.
Keywords: AMP-activated protein kinase; AMPK; CaMKK2; LKB1; biguanides; cancer; kinase activators; kinase inhibitors; tumour promoters; tumour suppressors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

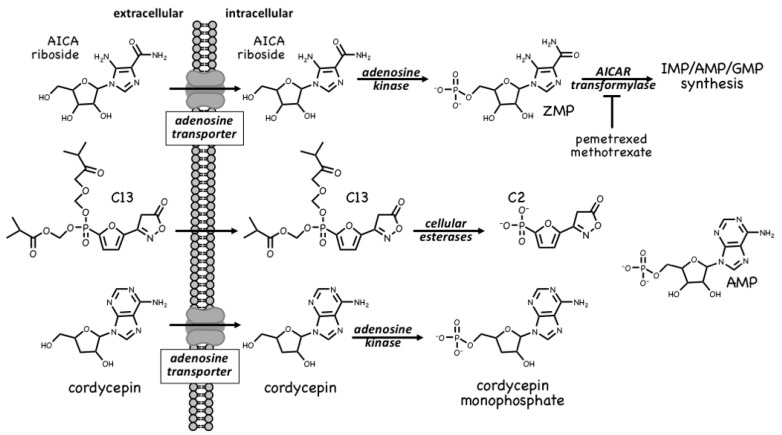
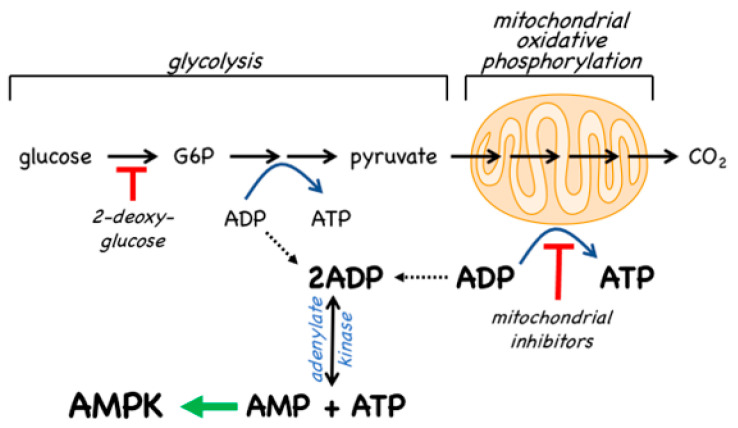
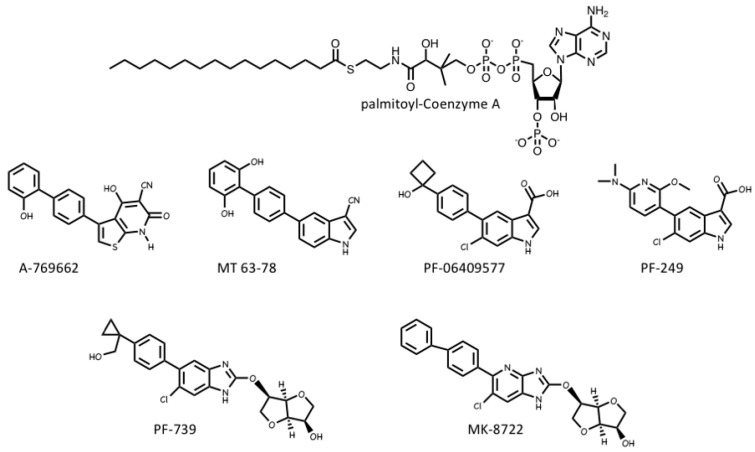
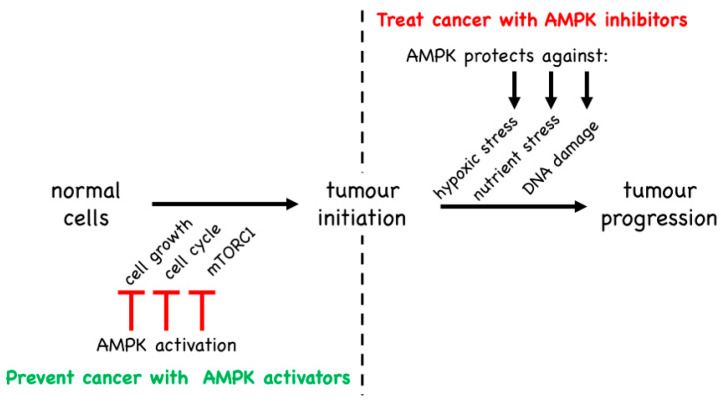
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
