

DNA polymerase ι compensates for Fanconi anemia pathway deficiency by countering DNA replication stress
- PMID: 33376220
- PMCID: PMC7777187
- DOI: 10.1073/pnas.2008821117
DNA polymerase ι compensates for Fanconi anemia pathway deficiency by countering DNA replication stress
Abstract
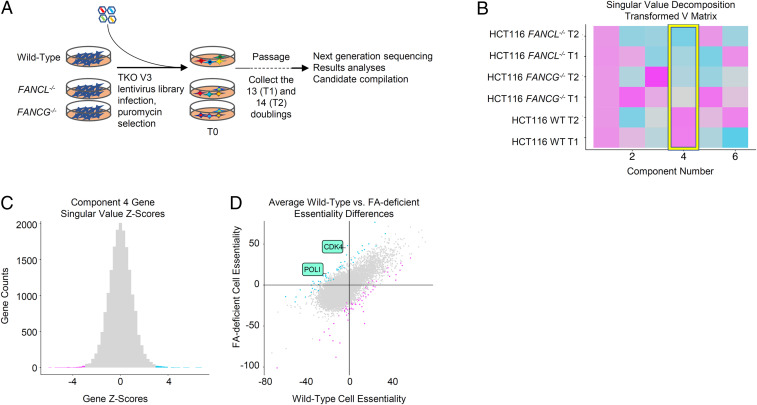
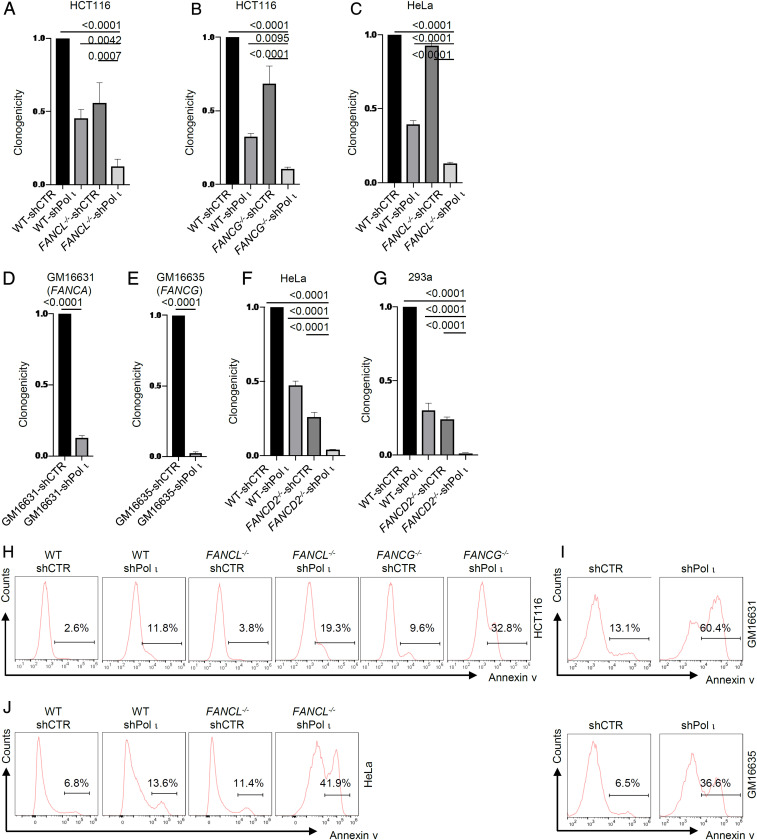
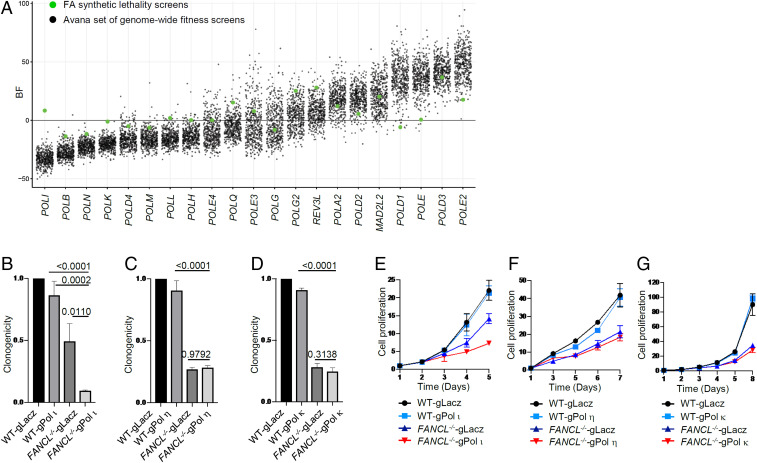
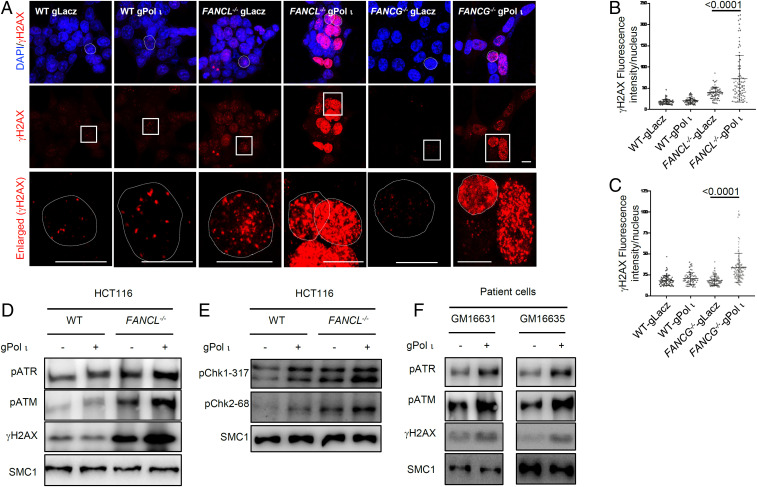
Fanconi anemia (FA) is caused by defects in cellular responses to DNA crosslinking damage and replication stress. Given the constant occurrence of endogenous DNA damage and replication fork stress, it is unclear why complete deletion of FA genes does not have a major impact on cell proliferation and germ-line FA patients are able to progress through development well into their adulthood. To identify potential cellular mechanisms that compensate for the FA deficiency, we performed dropout screens in FA mutant cells with a whole genome guide RNA library. This uncovered a comprehensive genome-wide profile of FA pathway synthetic lethality, including POLI and CDK4 As little is known of the cellular function of DNA polymerase iota (Pol ι), we focused on its role in the loss-of-function FA knockout mutants. Loss of both FA pathway function and Pol ι leads to synthetic defects in cell proliferation and cell survival, and an increase in DNA damage accumulation. Furthermore, FA-deficient cells depend on the function of Pol ι to resume replication upon replication fork stalling. Our results reveal a critical role for Pol ι in DNA repair and replication fork restart and suggest Pol ι as a target for therapeutic intervention in malignancies carrying an FA gene mutation.
Keywords: DNA polymerase; Fanconi anemia pathway; lesion bypass; whole genome fitness screens.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
The role of DNA polymerase iota in UV mutational spectra.Mutat Res. 2006 Jul 25;599(1-2):58-65. doi: 10.1016/j.mrfmmm.2006.01.003. Epub 2006 Feb 10. Mutat Res. 2006. PMID: 16472831
-
Polymerase iota (Pol ι) prevents PrimPol-mediated nascent DNA synthesis and chromosome instability.Sci Adv. 2023 Apr 14;9(15):eade7997. doi: 10.1126/sciadv.ade7997. Epub 2023 Apr 14. Sci Adv. 2023. PMID: 37058556 Free PMC article.
-
Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase iota.DNA Repair (Amst). 2003 Sep 18;2(9):991-1006. doi: 10.1016/s1568-7864(03)00094-6. DNA Repair (Amst). 2003. PMID: 12967656
-
Holding All the Cards-How Fanconi Anemia Proteins Deal with Replication Stress and Preserve Genomic Stability.Genes (Basel). 2019 Feb 22;10(2):170. doi: 10.3390/genes10020170. Genes (Basel). 2019. PMID: 30813363 Free PMC article. Review.
-
Fanconi anemia proteins and genome fragility: unraveling replication defects for cancer therapy.Trends Cancer. 2022 Jun;8(6):467-481. doi: 10.1016/j.trecan.2022.01.015. Epub 2022 Feb 26. Trends Cancer. 2022. PMID: 35232683 Review.
Cited by
-
Contracting eastern African C4 grasslands during the extinction of Paranthropus boisei.Sci Rep. 2021 Mar 30;11(1):7164. doi: 10.1038/s41598-021-86642-z. Sci Rep. 2021. PMID: 33785831 Free PMC article.
-
Fanconi Anemia Pathway Genes Advance Cervical Cancer via Immune Regulation and Cell Adhesion.Front Cell Dev Biol. 2021 Nov 15;9:734794. doi: 10.3389/fcell.2021.734794. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34869316 Free PMC article.
-
Implications of Translesion DNA Synthesis Polymerases on Genomic Stability and Human Health.Mol Cell Biol. 2023;43(8):401-425. doi: 10.1080/10985549.2023.2224199. Epub 2023 Jul 13. Mol Cell Biol. 2023. PMID: 37439479 Free PMC article.
-
E3 ubiquitin ligase RNF2 protects polymerase ι from destabilization.Biochim Biophys Acta Mol Cell Res. 2024 Jun;1871(5):119743. doi: 10.1016/j.bbamcr.2024.119743. Epub 2024 May 3. Biochim Biophys Acta Mol Cell Res. 2024. PMID: 38705361 Free PMC article.
-
Revolutionizing DNA repair research and cancer therapy with CRISPR-Cas screens.Nat Rev Mol Cell Biol. 2023 Jul;24(7):477-494. doi: 10.1038/s41580-022-00571-x. Epub 2023 Feb 13. Nat Rev Mol Cell Biol. 2023. PMID: 36781955 Review.
References
-
- Alter B. P., Fanconi’s anemia and malignancies. Am. J. Hematol. 53, 99–110 (1996). - PubMed
-
- D’Andrea A. D., Grompe M., The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 3, 23–34 (2003). - PubMed
-
- Xie Y., et al. , Aberrant Fanconi anaemia protein profiles in acute myeloid leukaemia cells. Br. J. Haematol. 111, 1057–1064 (2000). - PubMed
-
- Condie A., et al. , Analysis of the Fanconi anaemia complementation group A gene in acute myeloid leukaemia. Leuk. Lymphoma 43, 1849–1853 (2002). - PubMed
-
- van der Heijden M. S., Yeo C. J., Hruban R. H., Kern S. E., Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 63, 2585–2588 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
