

Ethnicity-related DMD Genotype Landscapes in European and Non-European Countries
- PMID: 33376799
- PMCID: PMC7768913
- DOI: 10.1212/NXG.0000000000000536
Ethnicity-related DMD Genotype Landscapes in European and Non-European Countries
Abstract
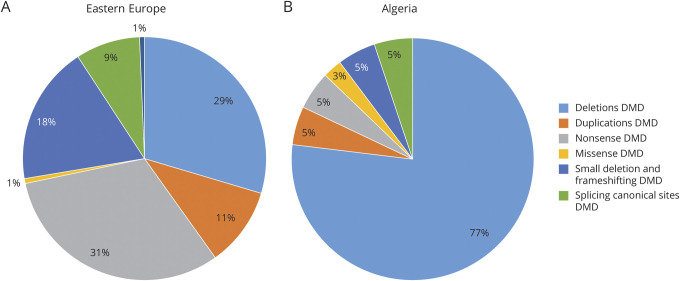
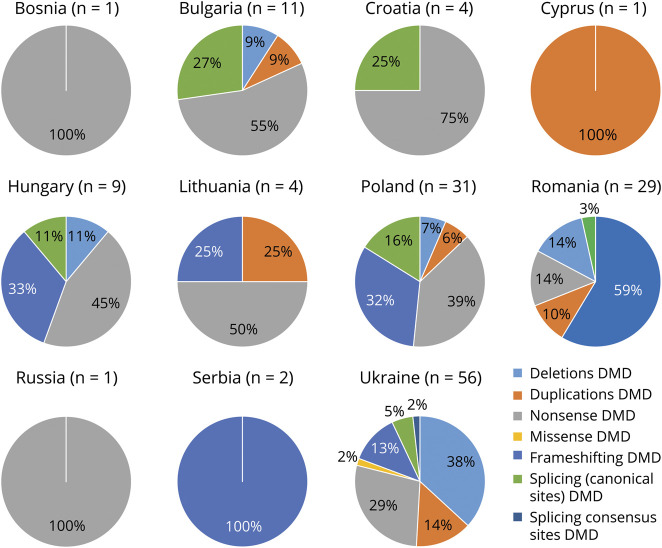
Objective: Genetic diagnosis and mutation identification are now compulsory for Duchenne (DMD) and Becker muscular dystrophies (BMD), which are due to dystrophin (DMD) gene mutations, either for disease prevention or personalized therapies. To evaluate the ethnic-related genetic assortments of DMD mutations, which may impact on DMD genetic diagnosis pipelines, we studied 328 patients with DMD and BMD from non-European countries.
Methods: We performed a full DMD mutation detection in 328 patients from 10 Eastern European countries (Poland, Hungary, Lithuania, Romania, Serbia, Croatia, Bosnia, Bulgaria, Ukraine, and Russia) and 2 non-European countries (Cyprus and Algeria). We used both conventional methods (multiplex ligation-dependent probe amplification [MLPA] followed by gene-specific sequencing) and whole-exome sequencing (WES) as a pivotal study ran in 28 patients where DMD mutations were already identified by standard techniques. WES output was also interrogated for DMD gene modifiers.
Results: We identified DMD gene mutations in 222 male patients. We identified a remarkable allele heterogeneity among different populations with a mutation landscape often country specific. We also showed that WES is effective for picking up all DMD deletions and small mutations and its adoption could allow a detection rate close to 90% of all occurring mutations. Gene modifiers haplotypes were identified with some ethnic-specific configurations.
Conclusions: Our data provide unreported mutation landscapes in different countries, suggesting that ethnicity may orient genetic diagnosis flowchart, which can be adjusted depending on the mutation type frequency, with impact in drug eligibility.
© 2020 American Academy of Neurology.
Figures
References
-
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–740. - PubMed
-
- Saengpattrachai M, Ray PN, Hawkins CE, Berzen A, Banwell BL. Grandpa and I have dystrophinopathy?: approach to asymptomatic hyperCKemia. Pediatr Neurol 2006;35:145–149. - PubMed
-
- Straub V, Balabanov P, Bushby K, et al. Stakeholder cooperation to overcome challenges in orphan medicine development: the example of Duchenne muscular dystrophy. Lancet Neurol 2016;15:882–890. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous