

mir15a/mir16-1 cluster and its novel targeting molecules negatively regulate cardiac hypertrophy
- PMID: 33377640
- PMCID: PMC7737755
- DOI: 10.1002/ctm2.242
mir15a/mir16-1 cluster and its novel targeting molecules negatively regulate cardiac hypertrophy
Erratum in
-
CORRIGENDUM.Clin Transl Med. 2021 Feb;11(2):e302. doi: 10.1002/ctm2.302. Clin Transl Med. 2021. PMID: 33635006 Free PMC article. No abstract available.
Abstract
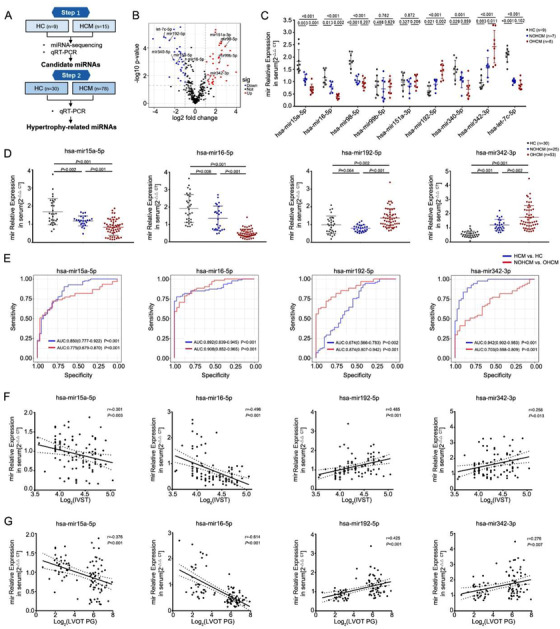
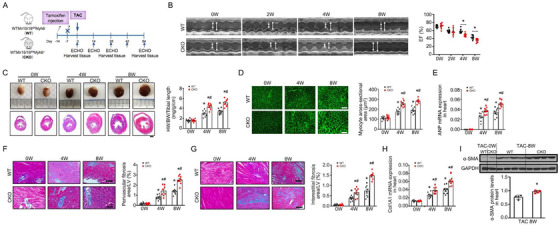
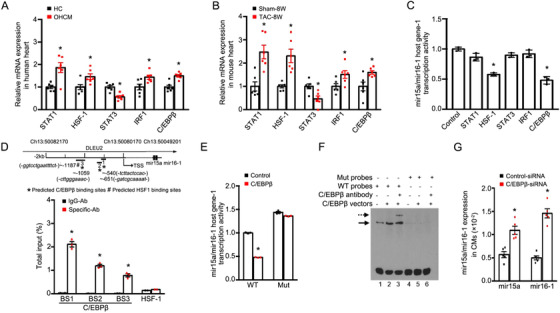
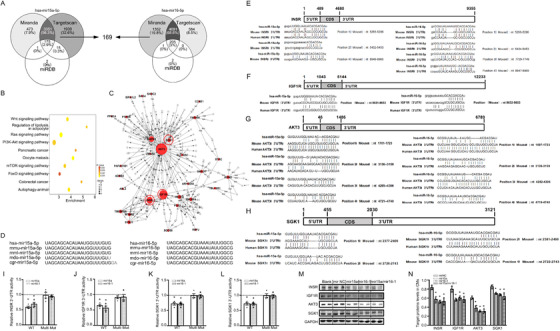
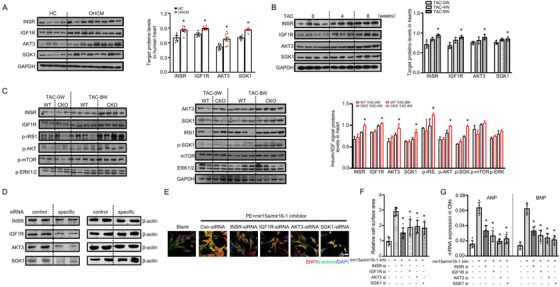
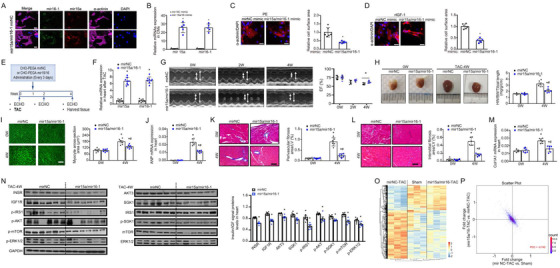
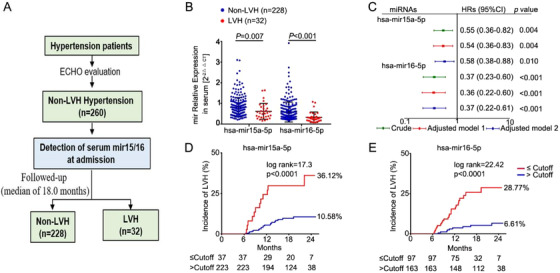
In response to pathological stimuli, the heart develops ventricular hypertrophy that progressively decompensates and leads to heart failure. miRNAs are increasingly recognized as pathogenic factors, clinically relevant biomarkers, and potential therapeutic targets. We identified that mir15a/mir16-1 cluster was negatively correlated with hypertrophic severity in patients with hypertrophic cardiomyopathy. The mir15a/mir16-1 expression was enriched in cardiomyocytes (CMs), decreased in hypertrophic human hearts, and decreased in mouse hearts after transverse aortic constriction (TAC). CM-specific mir15a/mir16-1 knockout promoted cardiac hypertrophy and dysfunction after TAC. CCAAT/enhancer binding protein (C/EBP)β was responsible for the downregulation of mir15a/mir16-1 cluster transcription. Mechanistically, mir15a/mir16-1 cluster attenuated the insulin/IGF1 signal transduction cascade by inhibiting multiple targets, including INSR, IGF-1R, AKT3, and serum/glucocorticoid regulated kinase 1 (SGK1). Pro-hypertrophic response induced by mir15a/mir16-1 inhibition was abolished by knockdown of insulin receptor (INSR), insulin like growth factor 1 receptor (IGF1R), AKT3, or SGK1. In vivo systemic delivery of mir15a/mir16-1 by nanoparticles inhibited the hypertrophic phenotype induced by TAC. Importantly, decreased serum mir15a/mir16-1 levels predicted the occurrence of left ventricular hypertrophy in a cohort of patients with hypertension. Therefore, mir15a/mir16-1 cluster is a promising therapeutic target and biomarker for cardiac hypertrophy.
Keywords: biomarkers; cardiac hypertrophy; heart failure; miRNAs; therapeutic target.
© 2020 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported
Figures
References
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589‐600. - PubMed
-
- Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370‐1380. - PubMed
-
- Selvetella G, Hirsch E, Notte A, Tarone G, Lembo G. Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence. Cardiovasc Res. 2004;63(3):373‐380. - PubMed
-
- Bisping E, Wakula P, Poteser M, Heinzel FR. Targeting cardiac hypertrophy: toward a causal heart failure therapy. J Cardiovasc Pharmacol. 2014;64(4):293‐305. - PubMed
-
- Vegter EL, van der Meer P, de Windt LJ, Pinto YM, Voors AA. MicroRNAs in heart failure: from biomarker to target for therapy. Eur J Heart Fail. 2016;18(5):457‐468. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous