

Hybrid methods for combined experimental and computational determination of protein structure
- PMID: 33380110
- PMCID: PMC7773420
- DOI: 10.1063/5.0026025
Hybrid methods for combined experimental and computational determination of protein structure
Abstract
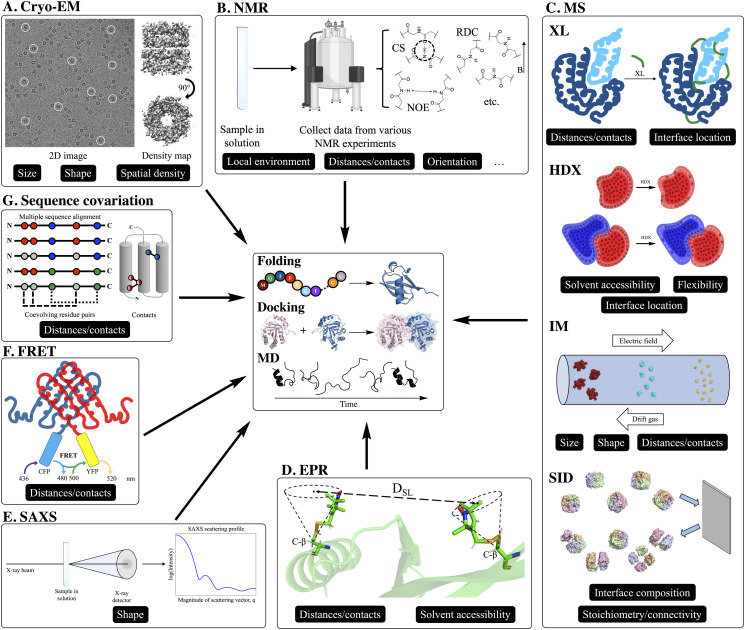
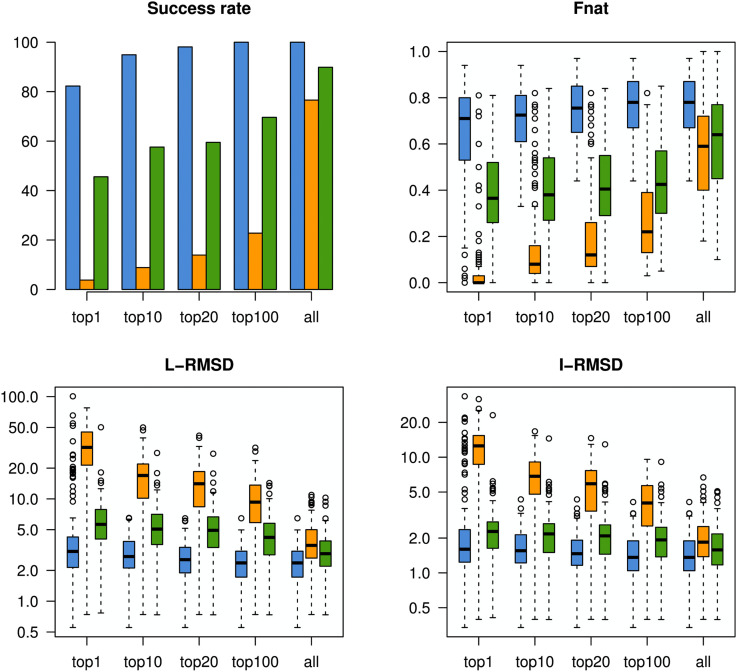
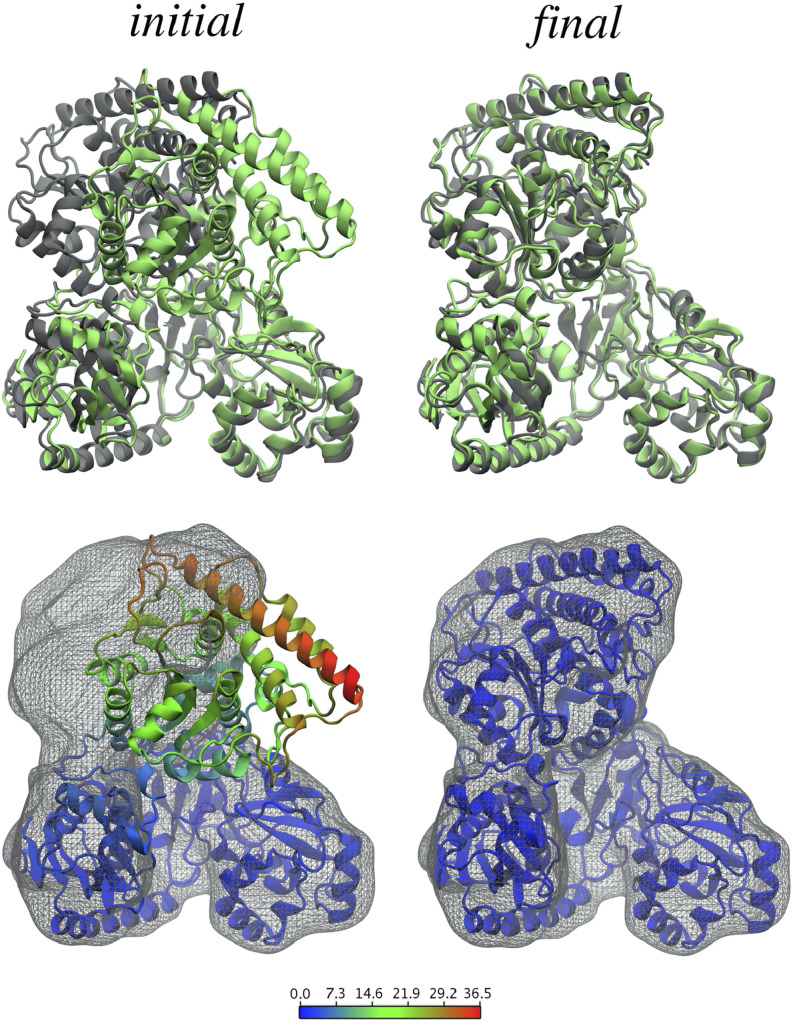
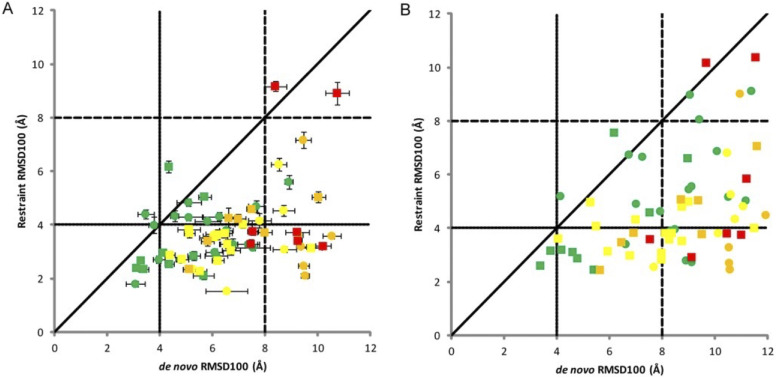
Knowledge of protein structure is paramount to the understanding of biological function, developing new therapeutics, and making detailed mechanistic hypotheses. Therefore, methods to accurately elucidate three-dimensional structures of proteins are in high demand. While there are a few experimental techniques that can routinely provide high-resolution structures, such as x-ray crystallography, nuclear magnetic resonance (NMR), and cryo-EM, which have been developed to determine the structures of proteins, these techniques each have shortcomings and thus cannot be used in all cases. However, additionally, a large number of experimental techniques that provide some structural information, but not enough to assign atomic positions with high certainty have been developed. These methods offer sparse experimental data, which can also be noisy and inaccurate in some instances. In cases where it is not possible to determine the structure of a protein experimentally, computational structure prediction methods can be used as an alternative. Although computational methods can be performed without any experimental data in a large number of studies, inclusion of sparse experimental data into these prediction methods has yielded significant improvement. In this Perspective, we cover many of the successes of integrative modeling, computational modeling with experimental data, specifically for protein folding, protein-protein docking, and molecular dynamics simulations. We describe methods that incorporate sparse data from cryo-EM, NMR, mass spectrometry, electron paramagnetic resonance, small-angle x-ray scattering, Förster resonance energy transfer, and genetic sequence covariation. Finally, we highlight some of the major challenges in the field as well as possible future directions.
Figures
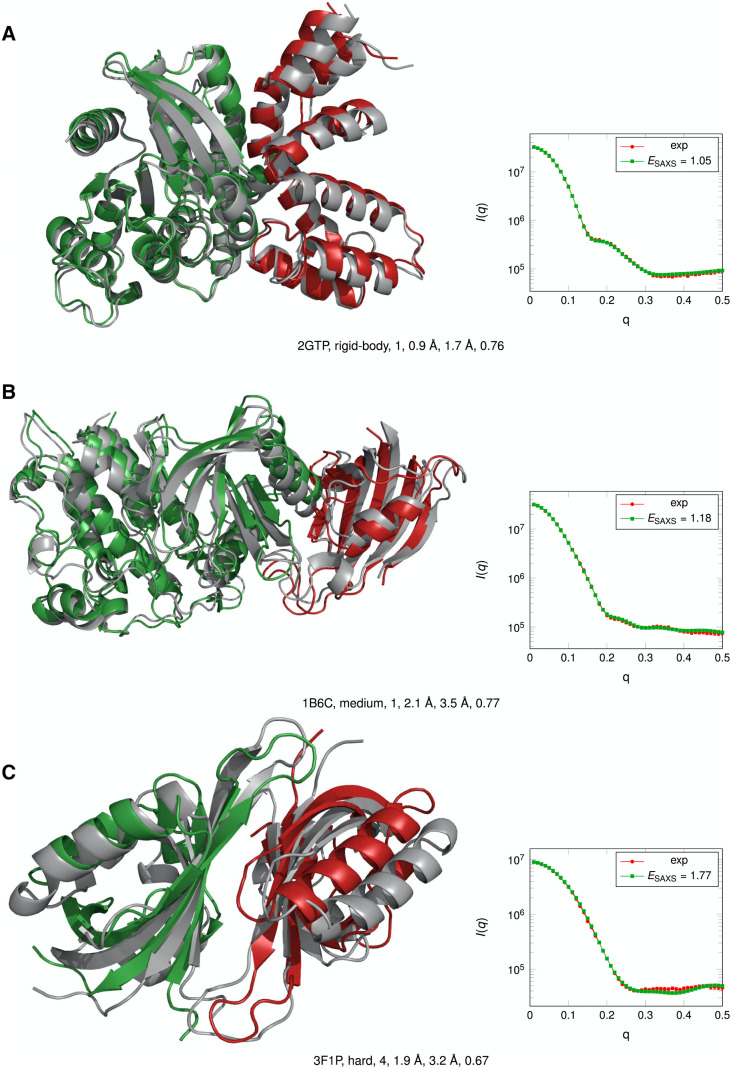
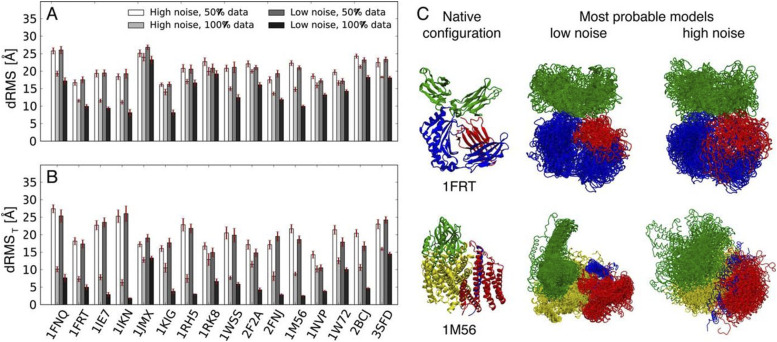
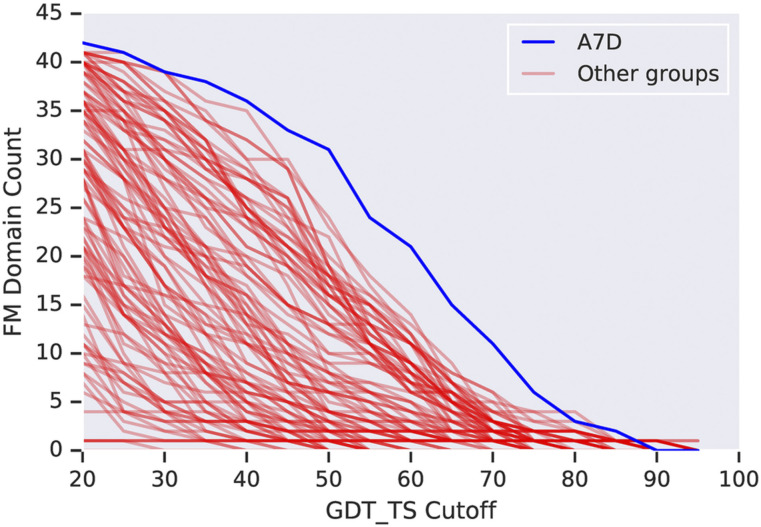
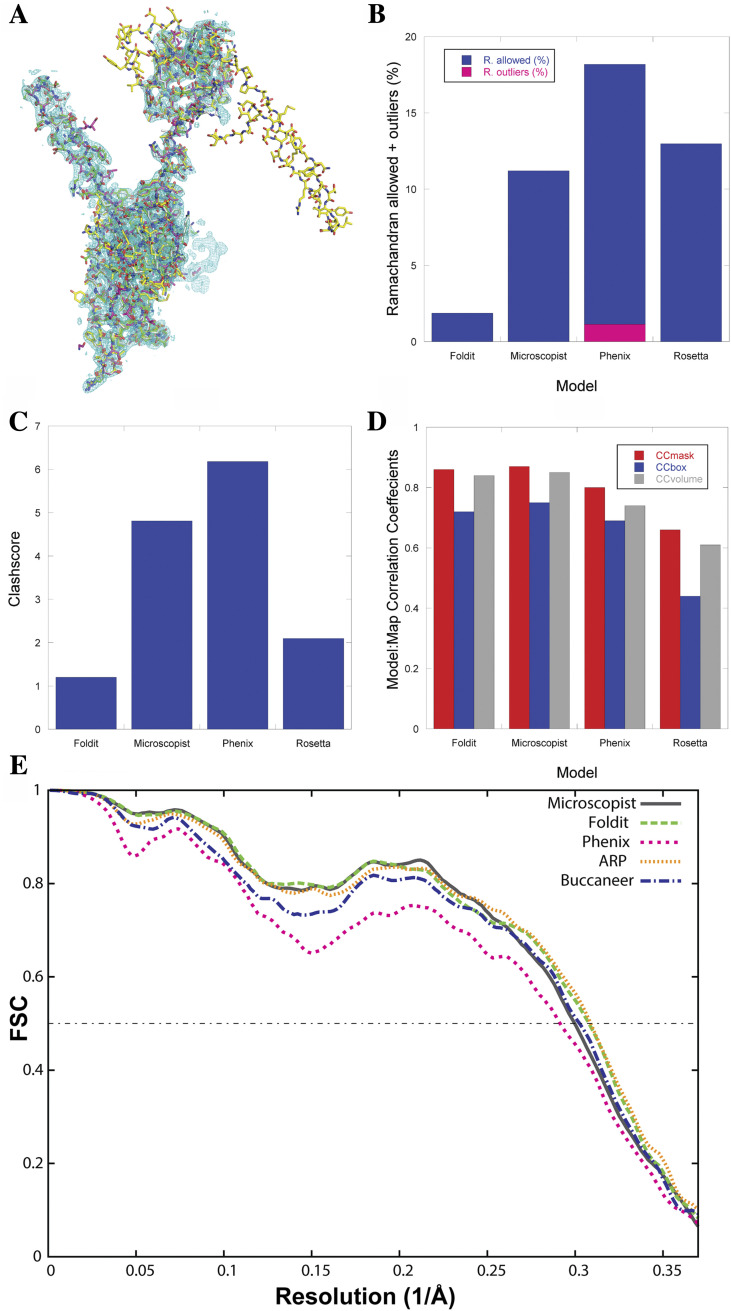
References
-
- Overall Growth of Released Structures per Year, RCSB, 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources