

Electronic Cigarettes Induce Mitochondrial DNA Damage and Trigger TLR9 (Toll-Like Receptor 9)-Mediated Atherosclerosis
- PMID: 33380174
- PMCID: PMC8608030
- DOI: 10.1161/ATVBAHA.120.315556
Electronic Cigarettes Induce Mitochondrial DNA Damage and Trigger TLR9 (Toll-Like Receptor 9)-Mediated Atherosclerosis
Abstract
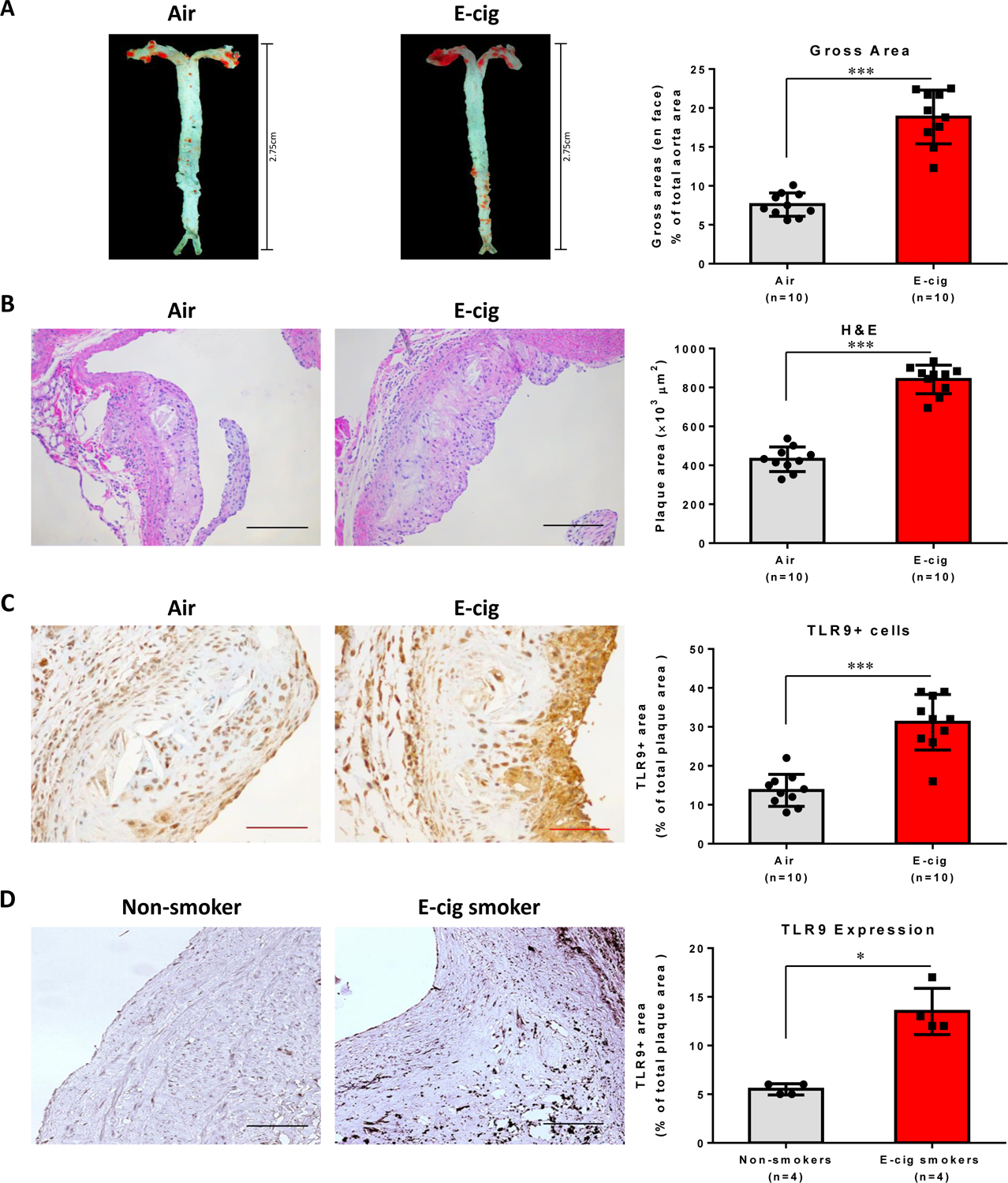
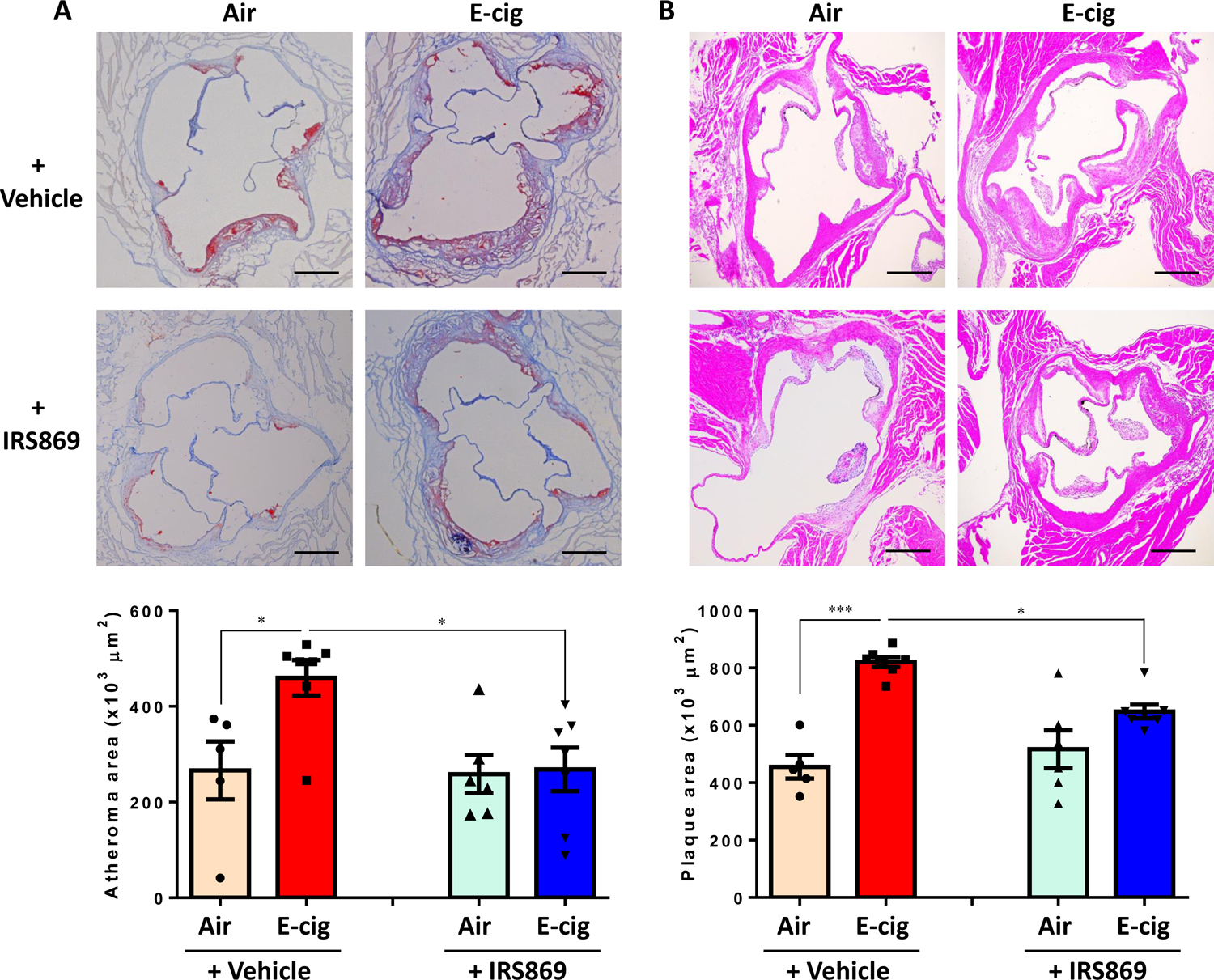
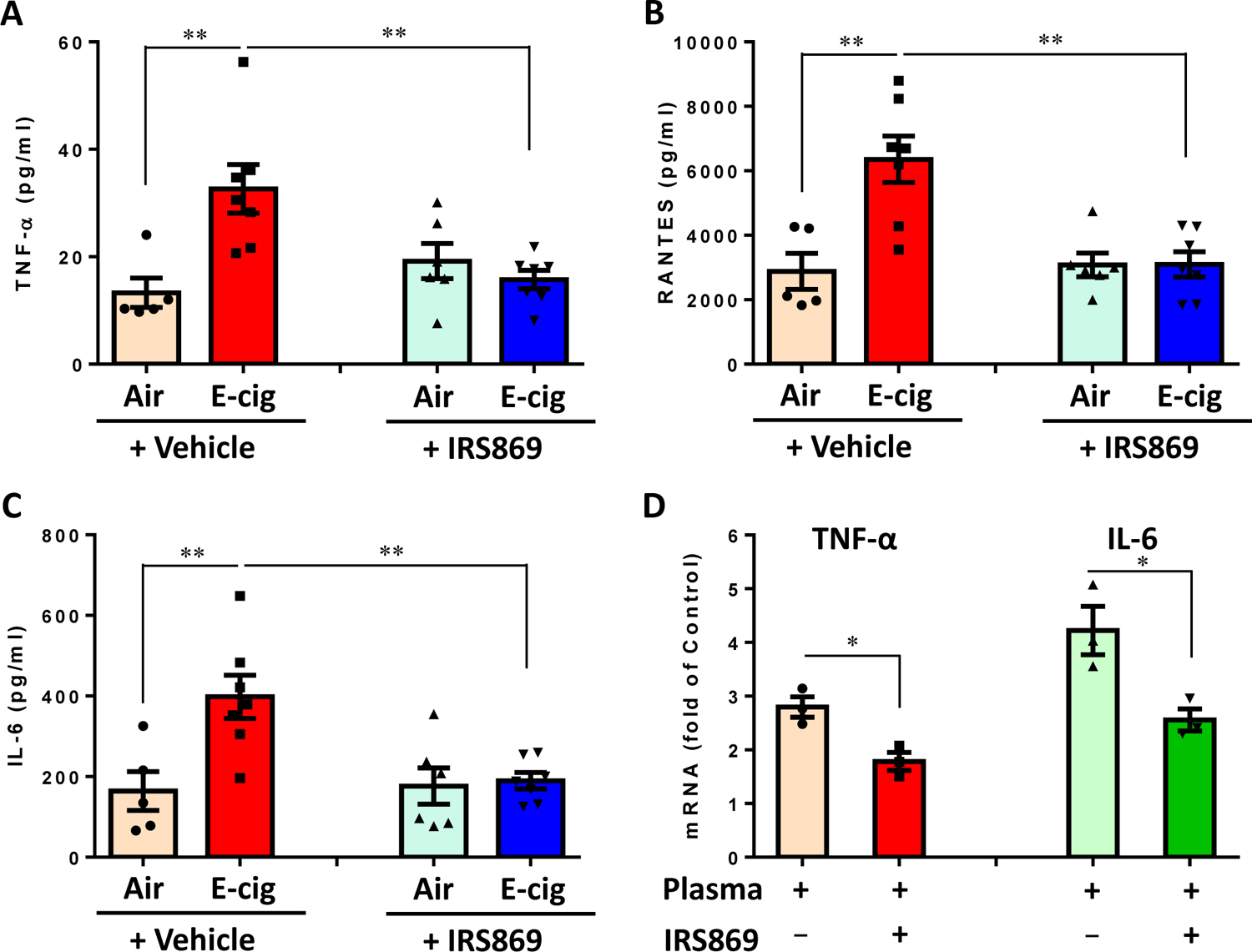
Objective: Electronic cigarette (e-cig) use has recently been implicated in promoting atherosclerosis. In this study, we aimed to investigate the mechanism of e-cig exposure accelerated atherosclerotic lesion development. Approach and Results: Eight-week-old ApoE-/- mice fed normal laboratory diet were exposed to e-cig vapor (ECV) for 2 hours/day, 5 days/week for 16 weeks. We found that ECV exposure significantly induced atherosclerotic lesions as examined by Oil Red O staining and greatly upregulated TLR9 (toll-like receptor 9) expression in classical monocytes and in the atherosclerotic plaques, which the latter was corroborated by enhanced TLR9 expression in human femoral artery atherosclerotic plaques from e-cig smokers. Intriguingly, we found a significant increase of oxidative mitochondria DNA lesion in the plasma of ECV-exposed mice. Administration of TLR9 antagonist before ECV exposure not only alleviated atherosclerosis and the upregulation of TLR9 in plaques but also attenuated the increase of plasma levels of inflammatory cytokines, reduced the plaque accumulation of lipid and macrophages, and decreased the frequency of blood CCR2+ (C-C chemokine receptor type 2) classical monocytes. Surprisingly, we found that cytoplasmic mitochondrial DNA isolated from ECV extract-treated macrophages can enhance TLR9 activation in reporter cells and the induction of inflammatory cytokine could be suppressed by TLR9 inhibitor in macrophages.
Conclusions: E-cig increases level of damaged mitochondrial DNA in circulating blood and induces the expression of TLR9, which elevate the expression of proinflammatory cytokines in monocyte/macrophage and consequently lead to atherosclerosis. Our results raise the possibility that intervention of TLR9 activation is a potential pharmacological target of ECV-related inflammation and cardiovascular diseases.
Keywords: atherosclerosis; cytokines; electronic cigarette; mice; monocyte.
Figures




References
-
- Ross R Atherosclerosis--an inflammatory disease. The New England journal of medicine. 1999;340:115–126 - PubMed
-
- Roth GA, Huffman MD, Moran AE, Feigin V, Mensah GA, Naghavi M, Murray CJ. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation. 2015;132:1667–1678 - PubMed
-
- Health. NCfCDPaHPUOoSa UA. The health consequences of smoking—50 years of progress: A report of the surgeon general. 2014
-
- Messner B, Bernhard D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:509–515 - PubMed
-
- Gairola CG, Drawdy ML, Block AE, Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein e−/− mice. Atherosclerosis. 2001;156:49–55 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
