

Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation
- PMID: 33380512
- PMCID: PMC8295506
- DOI: 10.1183/13993003.04229-2020
Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation
Abstract
Background: Heritable pulmonary arterial hypertension (PAH) is most commonly due to heterozygous mutations of the BMPR2 gene. Based on expert consensus, guidelines recommend annual screening echocardiography in asymptomatic BMPR2 mutation carriers. The main objectives of this study were to evaluate the characteristics of asymptomatic BMPR2 mutation carriers, assess their risk of occurrence of PAH and detect PAH at an early stage in this high-risk population.
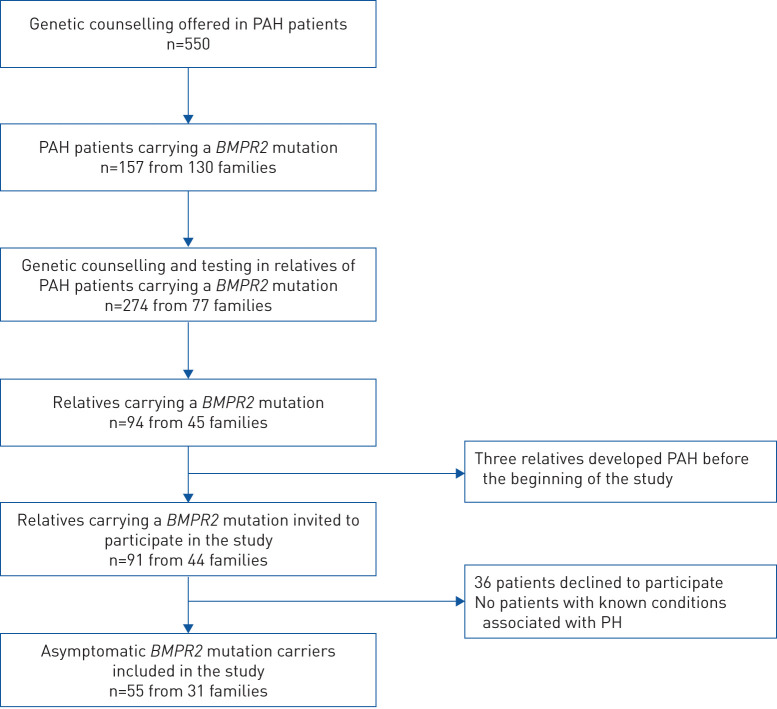
Methods: Asymptomatic BMPR2 mutation carriers underwent screening at baseline and annually for a minimum of 2 years (DELPHI-2 study; ClinicalTrials.gov: NCT01600898). Annual screening included clinical assessment, ECG, pulmonary function tests, 6-min walk distance, cardiopulmonary exercise testing, chest radiography, echocardiography and brain natriuretic peptide (BNP) or N-terminal (NT)-proBNP level. Right heart catheterisation (RHC) was performed based on predefined criteria. An optional RHC at rest and exercise was proposed at baseline.
Results: 55 subjects (26 males; median age 37 years) were included. At baseline, no PAH was suspected based on echocardiography and NT-proBNP levels. All subjects accepted RHC at inclusion, which identified two mild PAH cases (3.6%) and 12 subjects with exercise pulmonary hypertension (21.8%). At long-term follow-up (118.8 patient-years of follow-up), three additional cases were diagnosed, yielding a PAH incidence of 2.3% per year (0.99% per year in males and 3.5% per year in females). All PAH cases remained at low-risk status on oral therapy at last follow-up.
Conclusions: Asymptomatic BMPR2 mutation carriers have a significant risk of developing incident PAH. International multicentre studies are needed to confirm that refined multimodal screening programmes with regular follow-up allow early detection of PAH.
Copyright ©The authors 2021. For reproduction rights and permissions contact permissions@ersnet.org.
Conflict of interest statement
Conflict of interest: D. Montani reports grants and personal fees from Actelion and Bayer, personal fees from GlaxoSmithKline, Pfizer, Chiesi, Boehringer and Incyte Biosciences France, grants, personal fees and nonfinancial support from MSD, nonfinancial support from Acceleron, outside the submitted work. Conflict of interest: B. Girerd has nothing to disclose. Conflict of interest: X. Jaïs reports grants from Bayer, grants and personal fees from MSD, grants, personal fees and nonfinancial support from Actelion/Janssen, outside the submitted work. Conflict of interest: P. Laveneziana reports personal fees from Novartis France and Chiesi France, outside the submitted work. Conflict of interest: E.M.T. Lau reports grants and personal fees from Actelion and GlaxoSmithKline, outside the submitted work. Conflict of interest: A. Bouchachi has nothing to disclose. Conflict of interest: S. Hascoët reports grants and personal fees from Abbott, outside the submitted work. Conflict of interest: S. Gunther has nothing to disclose. Conflict of interest: L. Godinas has nothing to disclose. Conflict of interest: F. Parent has nothing to disclose. Conflict of interest: C. Guignabert has nothing to disclose. Conflict of interest: A. Beurnier has nothing to disclose. Conflict of interest: D. Chemla has nothing to disclose. Conflict of interest: P. Hervé has nothing to disclose. Conflict of interest: M. Eyries has nothing to disclose. Conflict of interest: F. Soubrier has nothing to disclose. Conflict of interest: G. Simonneau reports personal fees from Bayer and Acceleron, personal fees and nonfinancial support from MSD, outside the submitted work. Conflict of interest: O. Sitbon reports grants, personal fees and nonfinancial support from Actelion Pharmaceuticals, personal fees from Acceleron Pharmaceuticals, AOP Orphan, Ferrer and Gossamer Bio, grants and personal fees from Bayer HealthCare and MSD, grants from GlaxoSmithKline, outside the submitted work. Conflict of interest: L. Savale reports personal fees and nonfinancial support from Actelion and Bayer, grants, personal fees and nonfinancial support from GlaxoSmithKline, nonfinancial support from MSD, outside the submitted work. Conflict of interest: M. Humbert reports grants, personal fees and nonfinancial support from GlaxoSmithKline, personal fees from AstraZeneca, Novartis, Roche, Sanofi, Teva and Merck, grants and personal fees from Acceleron, Actelion and Bayer, outside the submitted work.
Figures




Comment in
-
Screening asymptomatic BMPR2 mutation carriers: a new frontier for pulmonary hypertension physicians?Eur Respir J. 2021 Jul 22;58(1):2100286. doi: 10.1183/13993003.00286-2021. Print 2021 Jul. Eur Respir J. 2021. PMID: 34301716 No abstract available.
References
-
- Galiè N, Humbert M, Vachiery J-L, et al. . 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Respir J 2015; 46: 903–975. doi:10.1183/13993003.01032-2015 - DOI - PubMed
-
- Galié N, Humbert M, Vachiery J-L, et al. . 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension – web addenda. The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and of the European Respiratory Society (ERS). Eur Heart J 2016; 37: 67–119. doi:10.1093/eurheartj/ehv317 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous