

Looking Back: A Short History of the Discovery of Enzymes and How They Became Powerful Chemical Tools
- PMID: 33381242
- PMCID: PMC7756376
- DOI: 10.1002/cctc.202001107
Looking Back: A Short History of the Discovery of Enzymes and How They Became Powerful Chemical Tools
Abstract
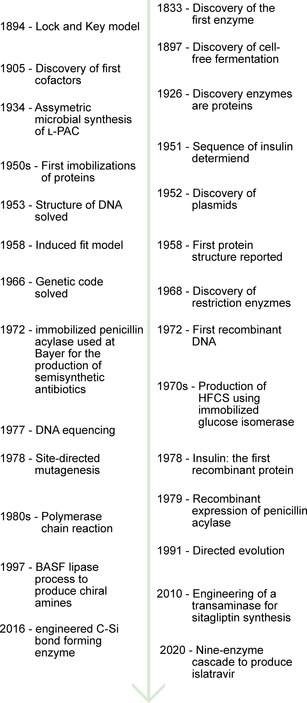
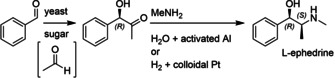
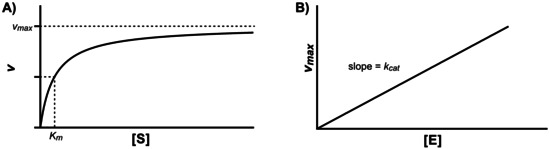
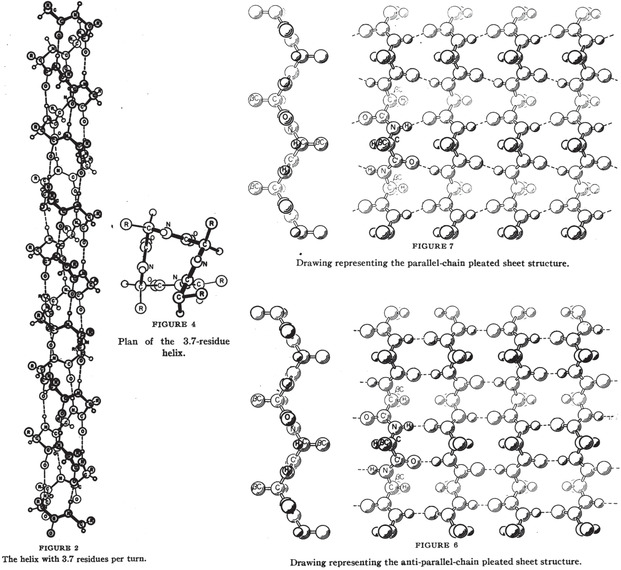
Enzymatic approaches to challenges in chemical synthesis are increasingly popular and very attractive to industry given their green nature and high efficiency compared to traditional methods. In this historical review we highlight the developments across several fields that were necessary to create the modern field of biocatalysis, with enzyme engineering and directed evolution at its core. We exemplify the modular, incremental, and highly unpredictable nature of scientific discovery, driven by curiosity, and showcase the resulting examples of cutting-edge enzymatic applications in industry.
Keywords: enzymes; green chemistry; history of biocatalysis; immobilization; protein engineering.
© 2020 The Authors. Published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
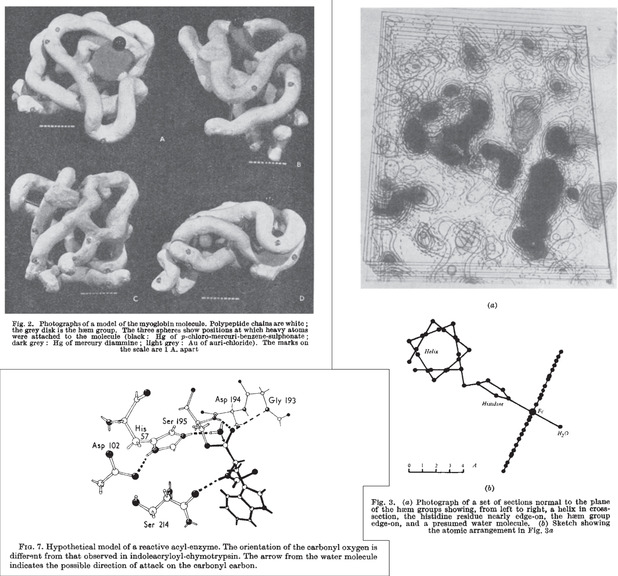
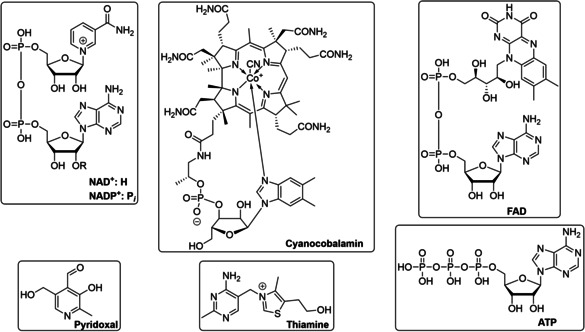
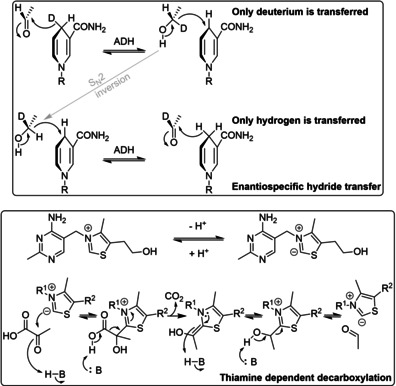
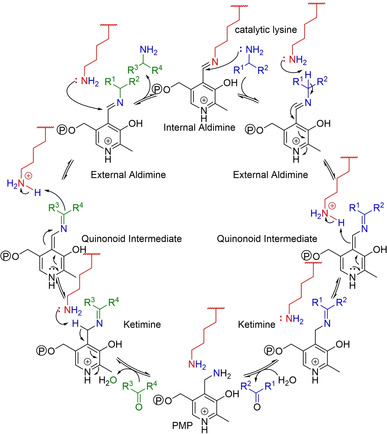
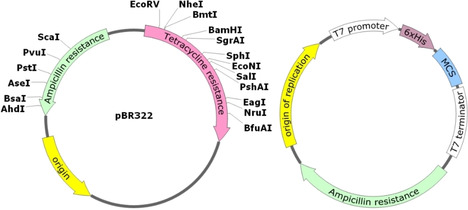
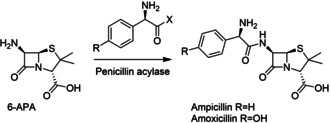
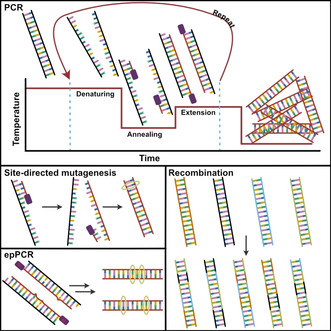

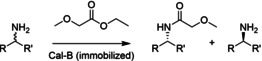
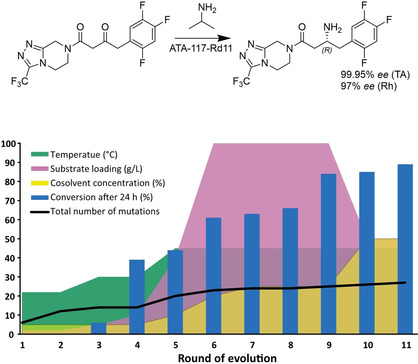
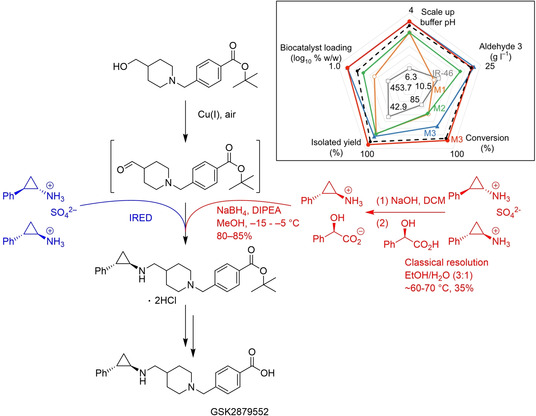
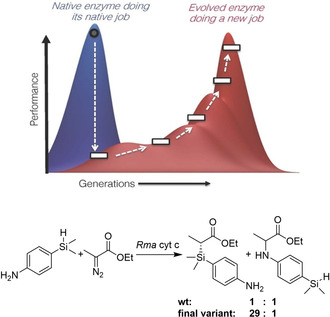
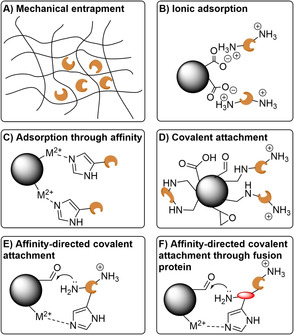
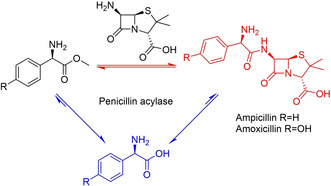
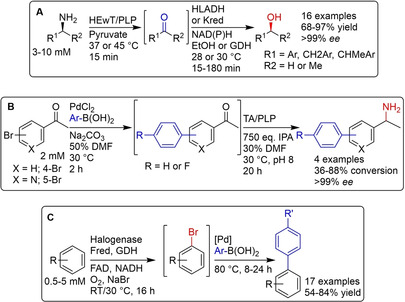
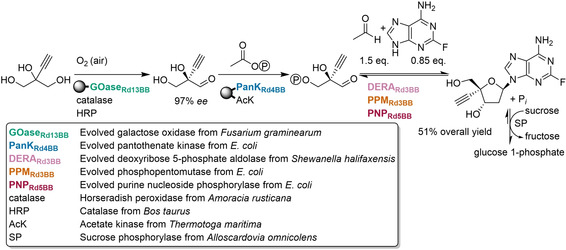
References
-
- Armstrong E. F., Nature 1933, 131, 535–537.
-
- E. Büchner, “Eduard Büchner – Nobel Lecture: Cell-free fermentation,” can be found under https://www.nobelprize.org/prizes/chemistry/1907/buchner/lecture/, 1907.
-
- W. Kühne, in Verhandlungen Des Naturhistorisch-Medicinischen Vereins Zu Heidelb, 1877, pp. 190–193.
-
- Robertson A. J. B., Platinum Met. Rev. 1975, 19, 64–69.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources