

PuMA: A papillomavirus genome annotation tool
- PMID: 33381306
- PMCID: PMC7751161
- DOI: 10.1093/ve/veaa068
PuMA: A papillomavirus genome annotation tool
Abstract
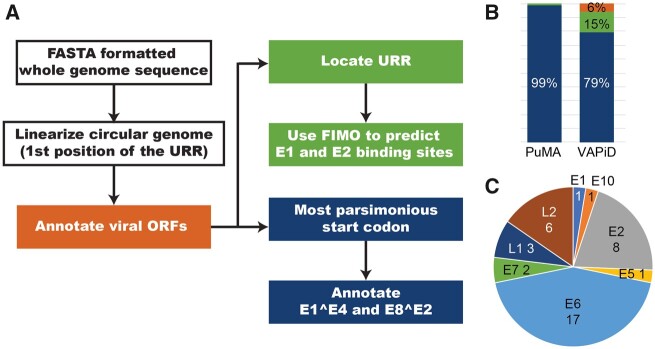
High-throughput sequencing technologies provide unprecedented power to identify novel viruses from a wide variety of (environmental) samples. The field of 'viral metagenomics' has dramatically expanded our understanding of viral diversity. Viral metagenomic approaches imply that many novel viruses will not be described by researchers who are experts on (the genomic organization of) that virus family. We have developed the papillomavirus annotation tool (PuMA) to provide researchers with a convenient and reproducible method to annotate and report novel papillomaviruses. PuMA currently correctly annotates 99% of the papillomavirus genes when benchmarked against the 655 reference genomes in the papillomavirus episteme. Compared to another viral annotation pipeline, PuMA annotates more viral features while being more accurate. To demonstrate its general applicability, we also developed a preliminary version of PuMA that can annotate polyomaviruses. PuMA is available on GitHub (https://github.com/KVD-lab/puma) and through the iMicrobe online environment (https://www.imicrobe.us/#/apps/puma).
Keywords: annotation; high-throughput sequencing; metagenomics; papillomavirus; polyomavirus; virome.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Altschul S. F. et al. (1990) ‘Basic Local Alignment Search Tool’, Journal of Molecular Biology, 215: 403–10. - PubMed
-
- Bailey T. L., Elkan C. (1994) ‘Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers’, Proceedings International Conference on Intelligent Systems for Molecular Biology, 2: 28–36. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
