

APOD: accurate sequence-based predictor of disordered flexible linkers
- PMID: 33381830
- PMCID: PMC7773485
- DOI: 10.1093/bioinformatics/btaa808
APOD: accurate sequence-based predictor of disordered flexible linkers
Abstract
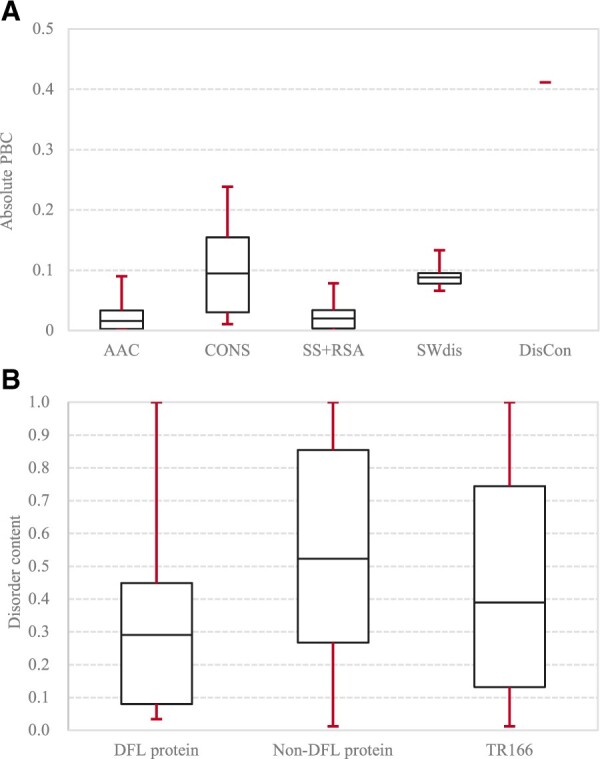
Motivation: Disordered flexible linkers (DFLs) are abundant and functionally important intrinsically disordered regions that connect protein domains and structural elements within domains and which facilitate disorder-based allosteric regulation. Although computational estimates suggest that thousands of proteins have DFLs, they were annotated experimentally in <200 proteins. This substantial annotation gap can be reduced with the help of accurate computational predictors. The sole predictor of DFLs, DFLpred, trade-off accuracy for shorter runtime by excluding relevant but computationally costly predictive inputs. Moreover, it relies on the local/window-based information while lacking to consider useful protein-level characteristics.
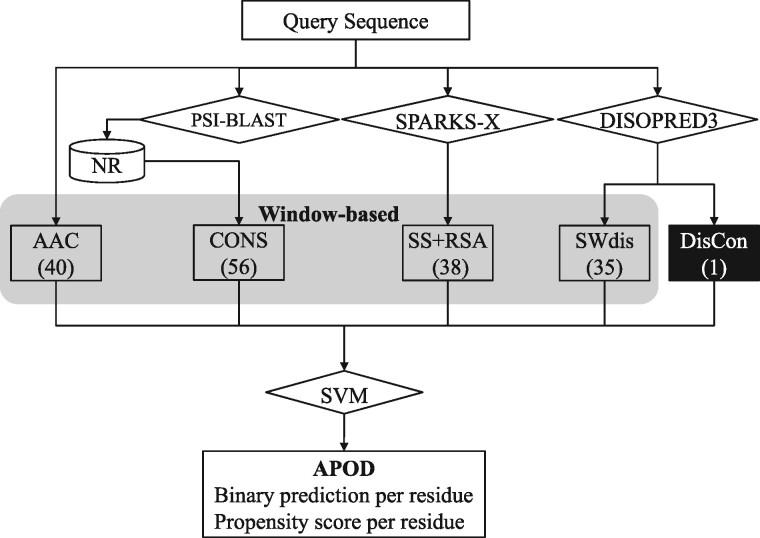
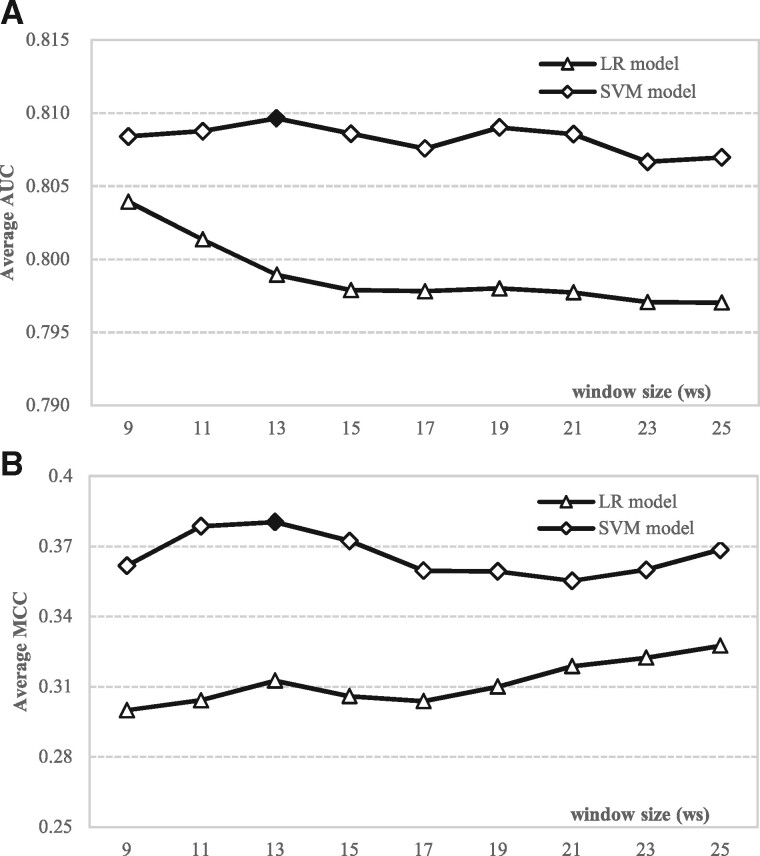
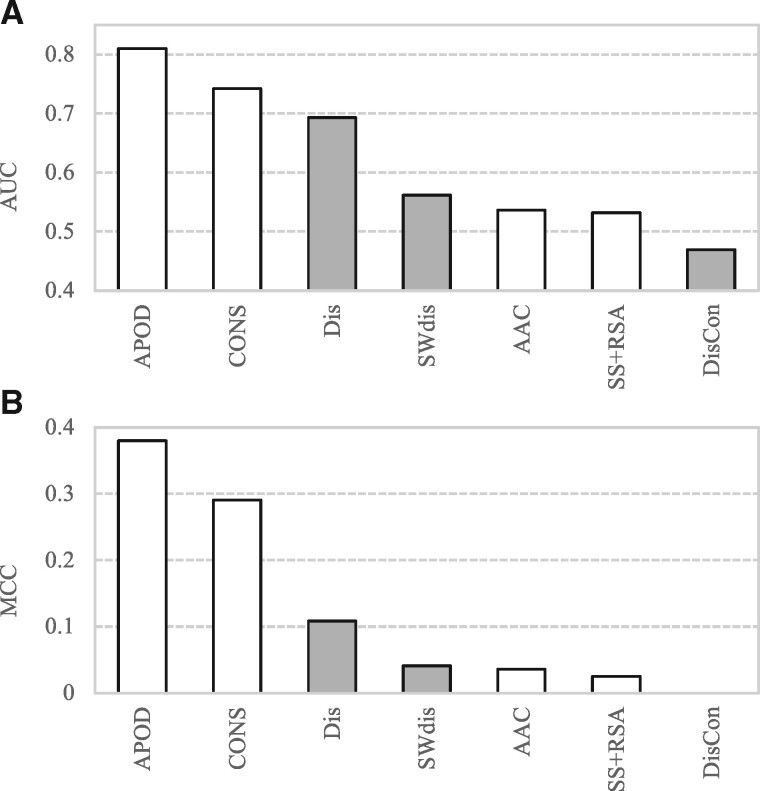
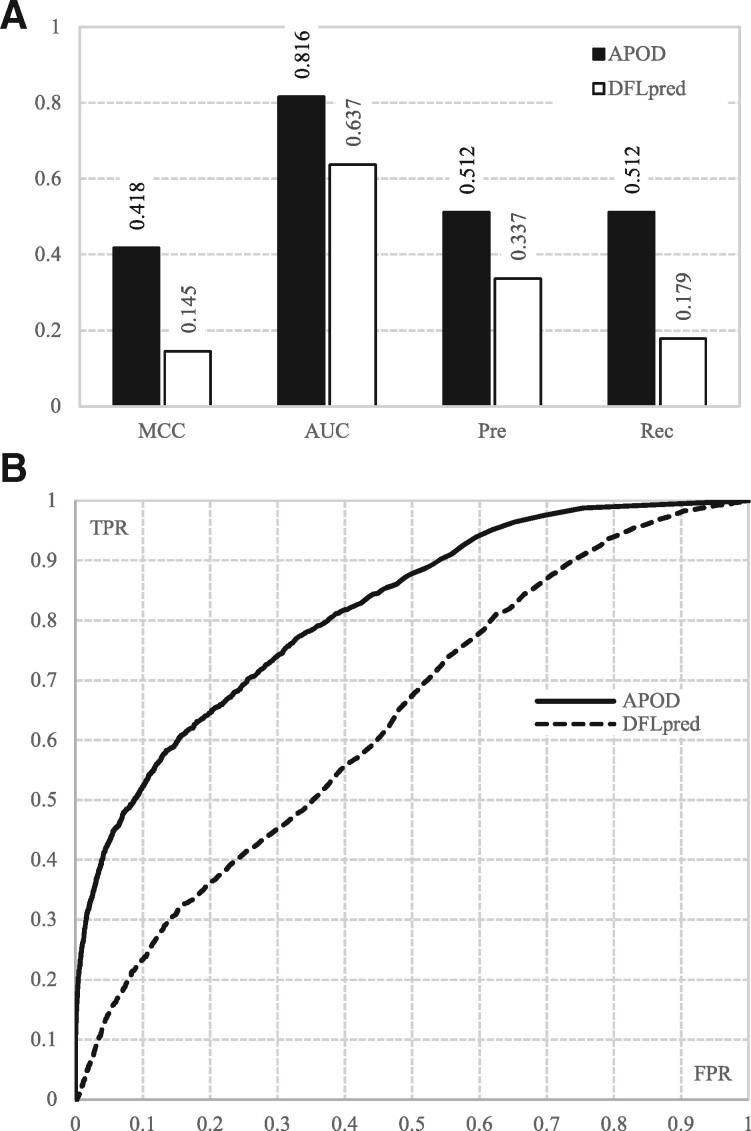
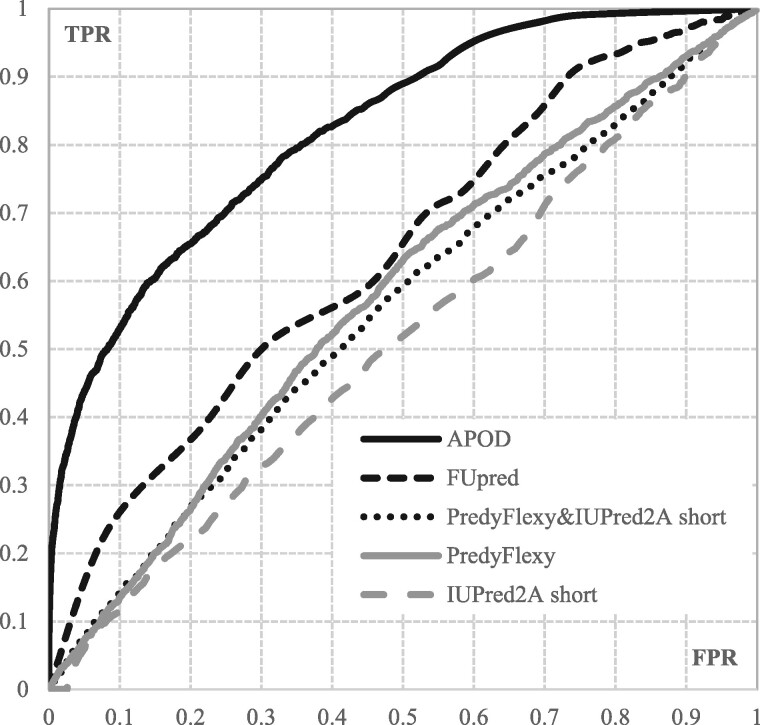
Results: We conceptualize, design and test APOD (Accurate Predictor Of DFLs), the first highly accurate predictor that utilizes both local- and protein-level inputs that quantify propensity for disorder, sequence composition, sequence conservation and selected putative structural properties. Consequently, APOD offers significantly more accurate predictions when compared with its faster predecessor, DFLpred, and several other alternative ways to predict DFLs. These improvements stem from the use of a more comprehensive set of inputs that cover the protein-level information and the application of a more sophisticated predictive model, a well-parametrized support vector machine. APOD achieves area under the curve = 0.82 (28% improvement over DFLpred) and Matthews correlation coefficient = 0.42 (180% increase over DFLpred) when tested on an independent/low-similarity test dataset. Consequently, APOD is a suitable choice for accurate and small-scale prediction of DFLs.
Availability and implementation: https://yanglab.nankai.edu.cn/APOD/.
© The Author(s) 2020. Published by Oxford University Press.
Figures
Similar articles
-
Prediction of Disordered Linkers Using APOD.Methods Mol Biol. 2025;2867:219-231. doi: 10.1007/978-1-0716-4196-5_13. Methods Mol Biol. 2025. PMID: 39576584
-
TransDFL: Identification of Disordered Flexible Linkers in Proteins by Transfer Learning.Genomics Proteomics Bioinformatics. 2023 Apr;21(2):359-369. doi: 10.1016/j.gpb.2022.10.004. Epub 2022 Oct 19. Genomics Proteomics Bioinformatics. 2023. PMID: 36272675 Free PMC article.
-
DFLpred: High-throughput prediction of disordered flexible linker regions in protein sequences.Bioinformatics. 2016 Jun 15;32(12):i341-i350. doi: 10.1093/bioinformatics/btw280. Bioinformatics. 2016. PMID: 27307636 Free PMC article.
-
Computational prediction of functions of intrinsically disordered regions.Prog Mol Biol Transl Sci. 2019;166:341-369. doi: 10.1016/bs.pmbts.2019.04.006. Epub 2019 May 20. Prog Mol Biol Transl Sci. 2019. PMID: 31521235 Review.
-
Accuracy of protein-level disorder predictions.Brief Bioinform. 2020 Sep 25;21(5):1509-1522. doi: 10.1093/bib/bbz100. Brief Bioinform. 2020. PMID: 31616935 Review.
Cited by
-
IDP-LM: Prediction of protein intrinsic disorder and disorder functions based on language models.PLoS Comput Biol. 2023 Nov 22;19(11):e1011657. doi: 10.1371/journal.pcbi.1011657. eCollection 2023 Nov. PLoS Comput Biol. 2023. PMID: 37992088 Free PMC article.
-
flDPnn: Accurate intrinsic disorder prediction with putative propensities of disorder functions.Nat Commun. 2021 Jul 21;12(1):4438. doi: 10.1038/s41467-021-24773-7. Nat Commun. 2021. PMID: 34290238 Free PMC article.
-
CAID prediction portal: a comprehensive service for predicting intrinsic disorder and binding regions in proteins.Nucleic Acids Res. 2023 Jul 5;51(W1):W62-W69. doi: 10.1093/nar/gkad430. Nucleic Acids Res. 2023. PMID: 37246642 Free PMC article.
-
Prediction of Disordered Linkers Using APOD.Methods Mol Biol. 2025;2867:219-231. doi: 10.1007/978-1-0716-4196-5_13. Methods Mol Biol. 2025. PMID: 39576584
-
TransDFL: Identification of Disordered Flexible Linkers in Proteins by Transfer Learning.Genomics Proteomics Bioinformatics. 2023 Apr;21(2):359-369. doi: 10.1016/j.gpb.2022.10.004. Epub 2022 Oct 19. Genomics Proteomics Bioinformatics. 2023. PMID: 36272675 Free PMC article.
References
-
- Anand S., Mohanty D. (2012) Inter-domain movements in polyketide synthases: a molecular dynamics study. Mol. Biosyst., 8, 1157–1171. - PubMed
-
- Arbesu M., Pons M. (2019) Integrating disorder in globular multidomain proteins: fuzzy sensors and the role of SH3 domains. Arch. Biochem. Biophys., 677, 108161. - PubMed
-
- Atas H. et al. (2018) Phylogenetic and other conservation-based approaches to predict protein functional sites. Methods Mol. Biol., 1762, 51–69. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
