

The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants
- PMID: 33382980
- PMCID: PMC7878430
- DOI: 10.1016/j.chom.2020.12.001
The cancer microbiome atlas: a pan-cancer comparative analysis to distinguish tissue-resident microbiota from contaminants
Abstract
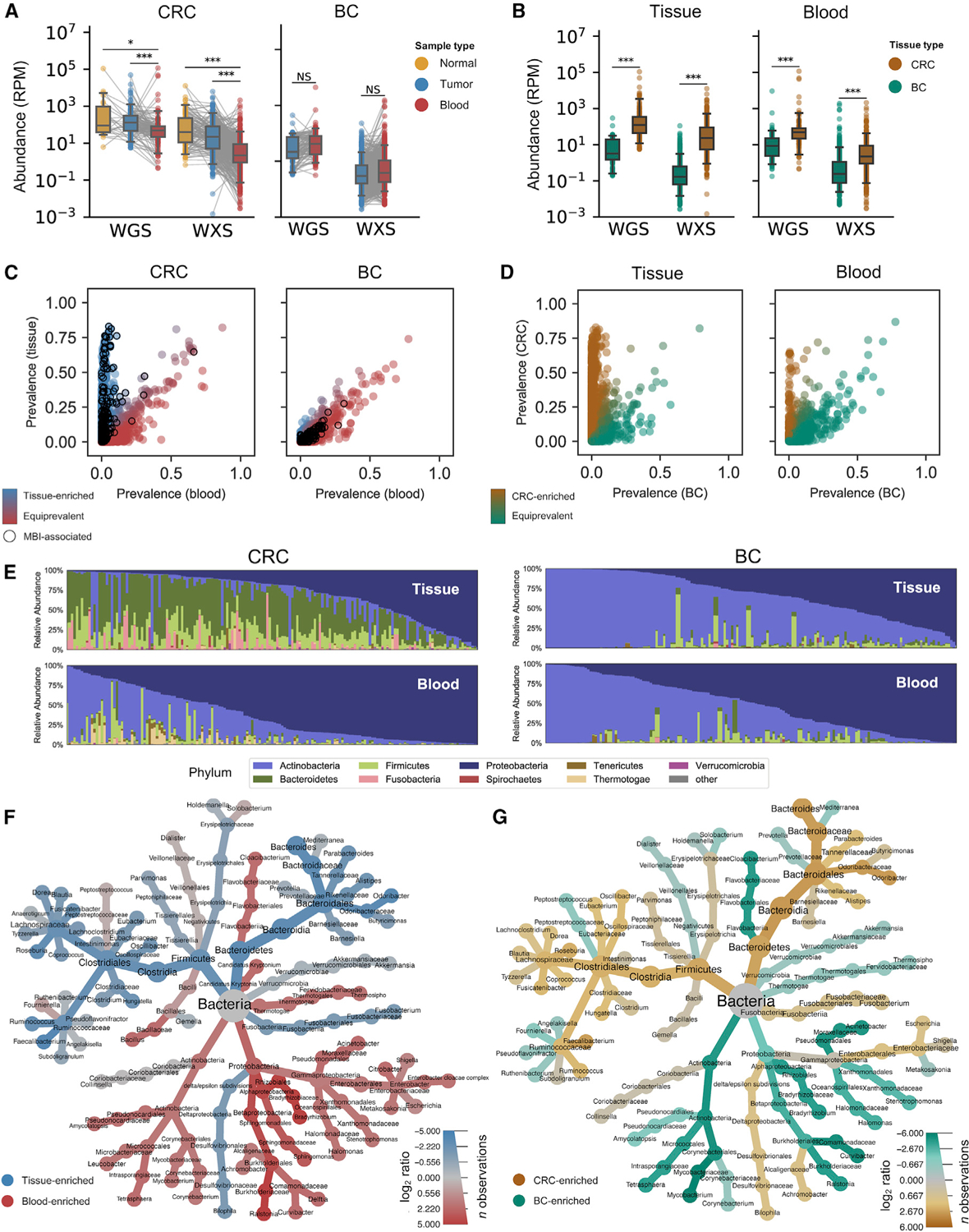
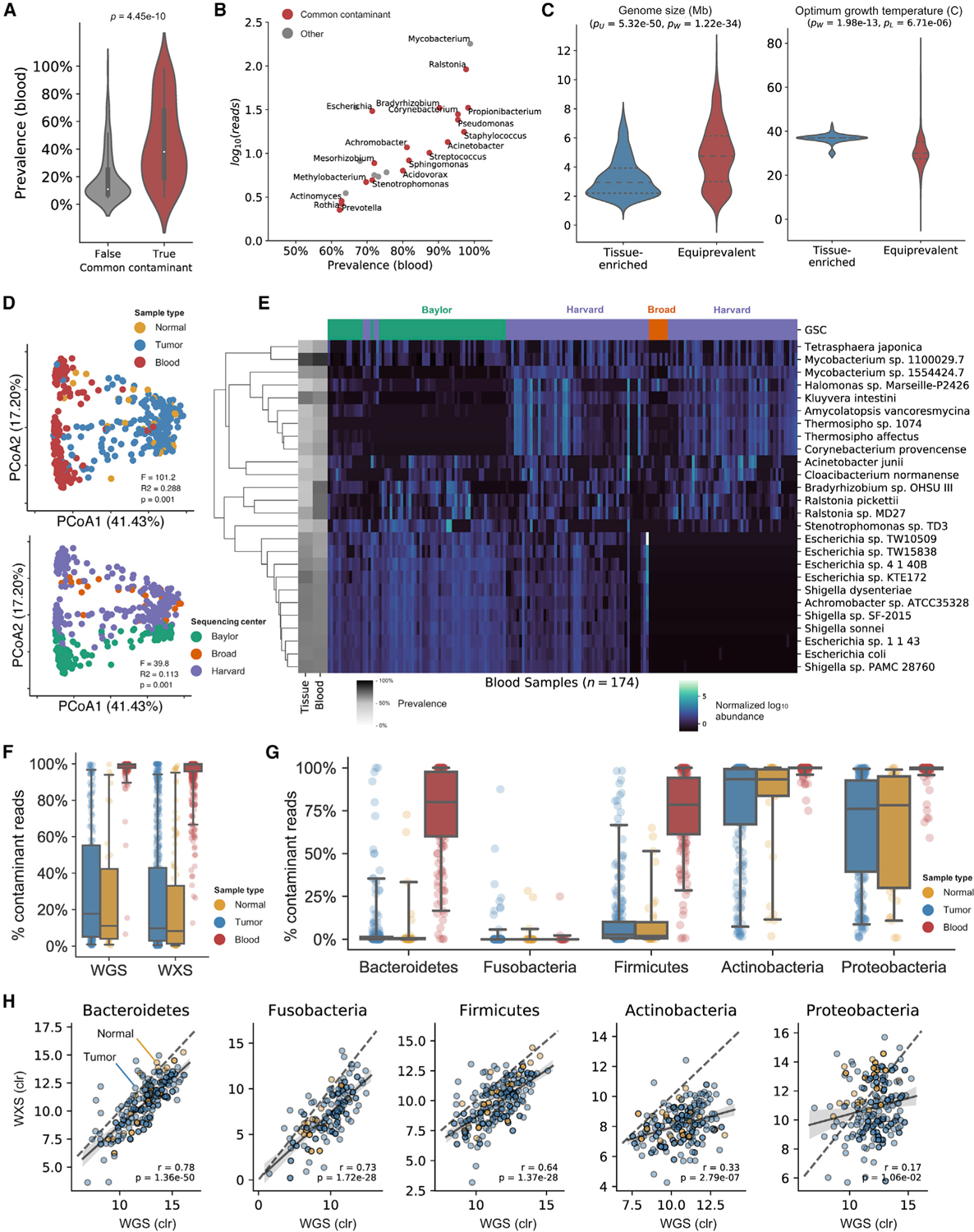
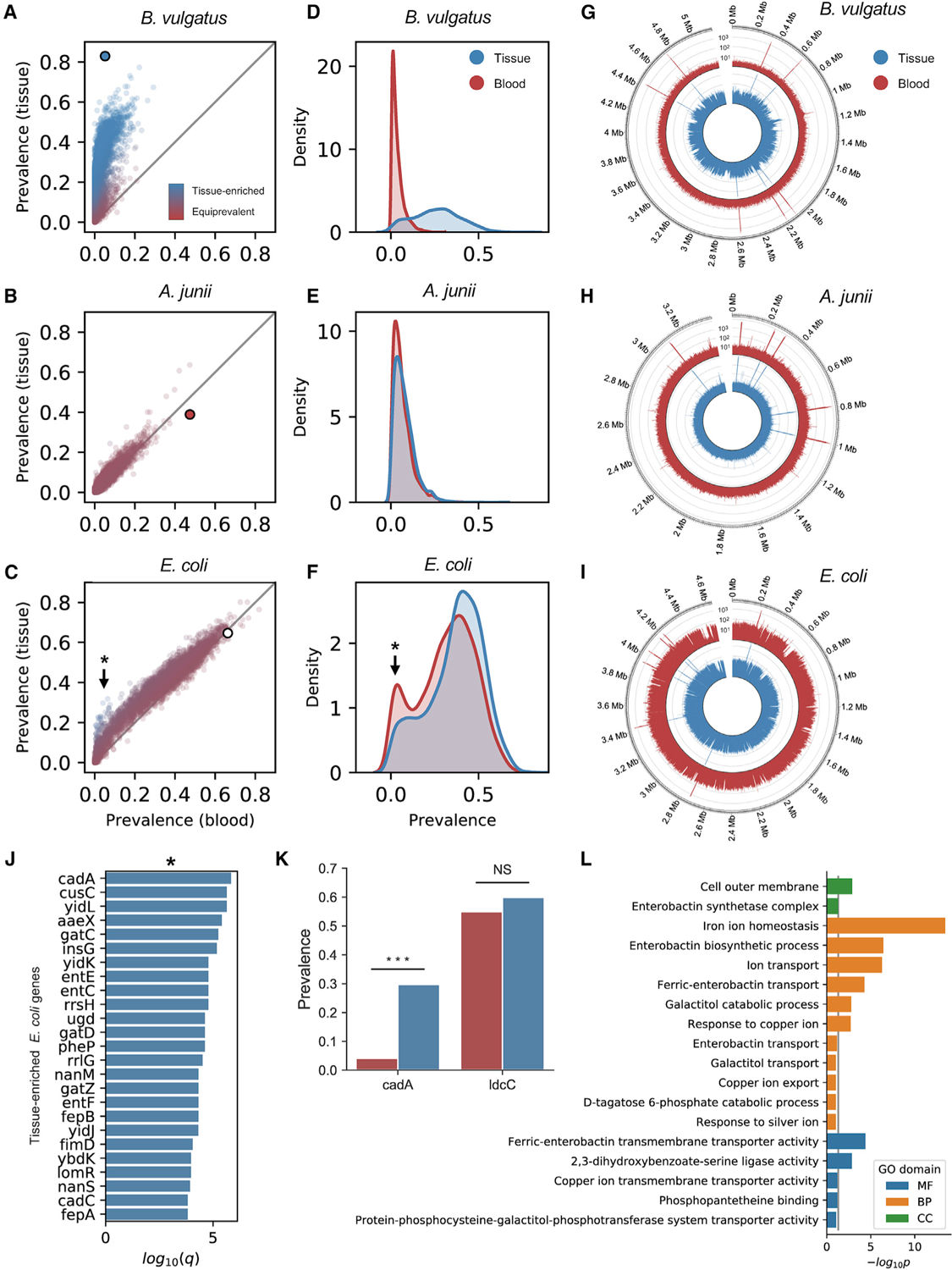
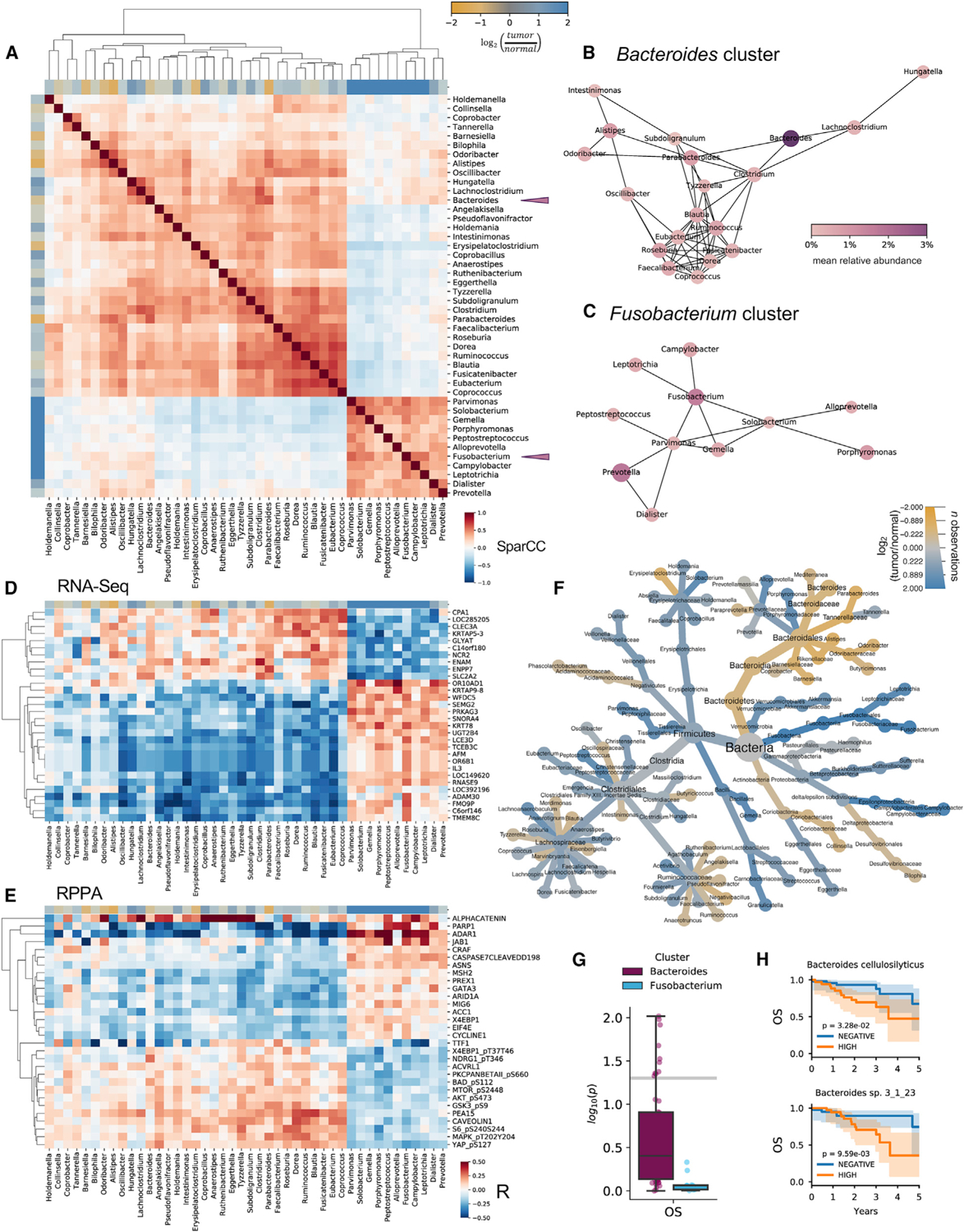
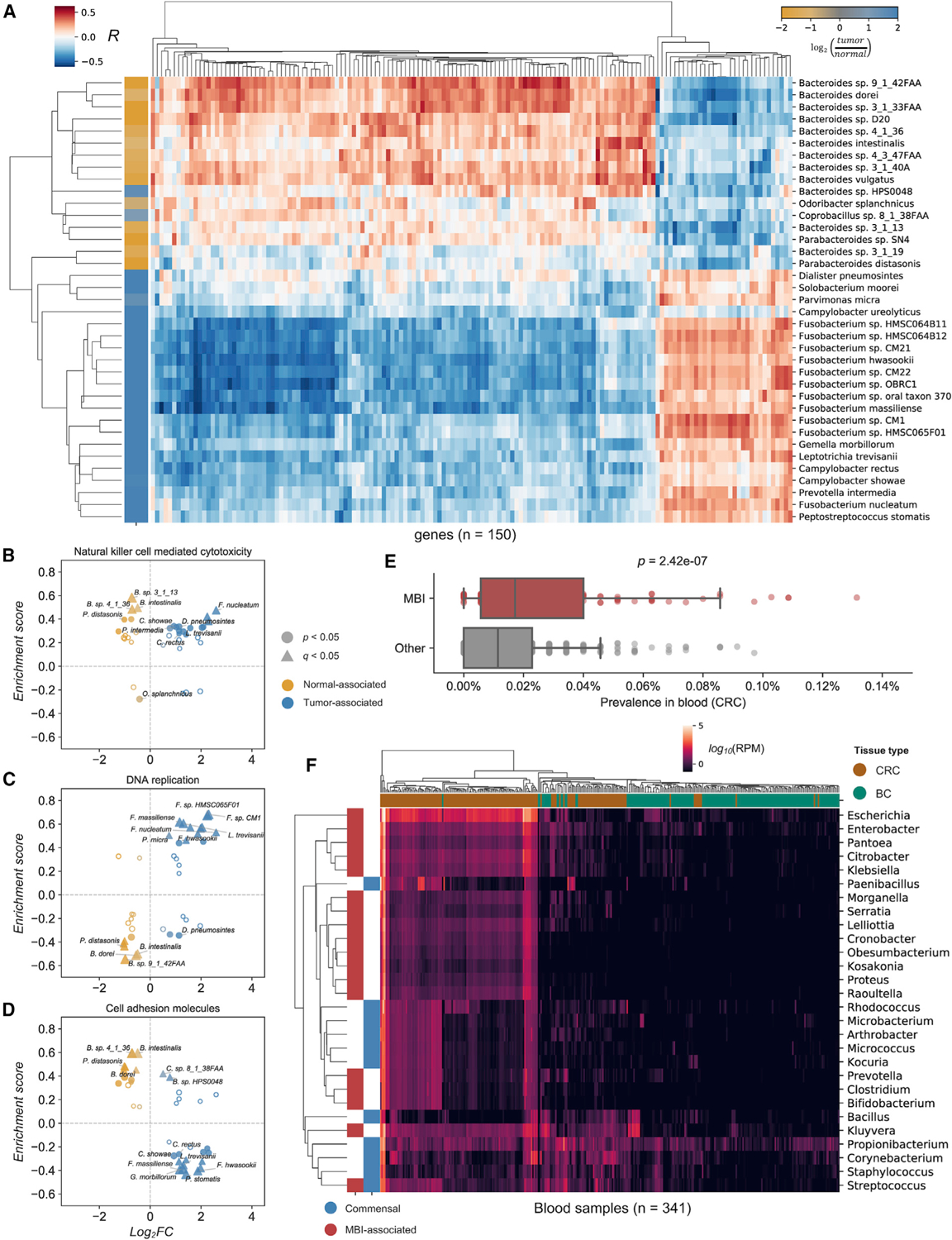
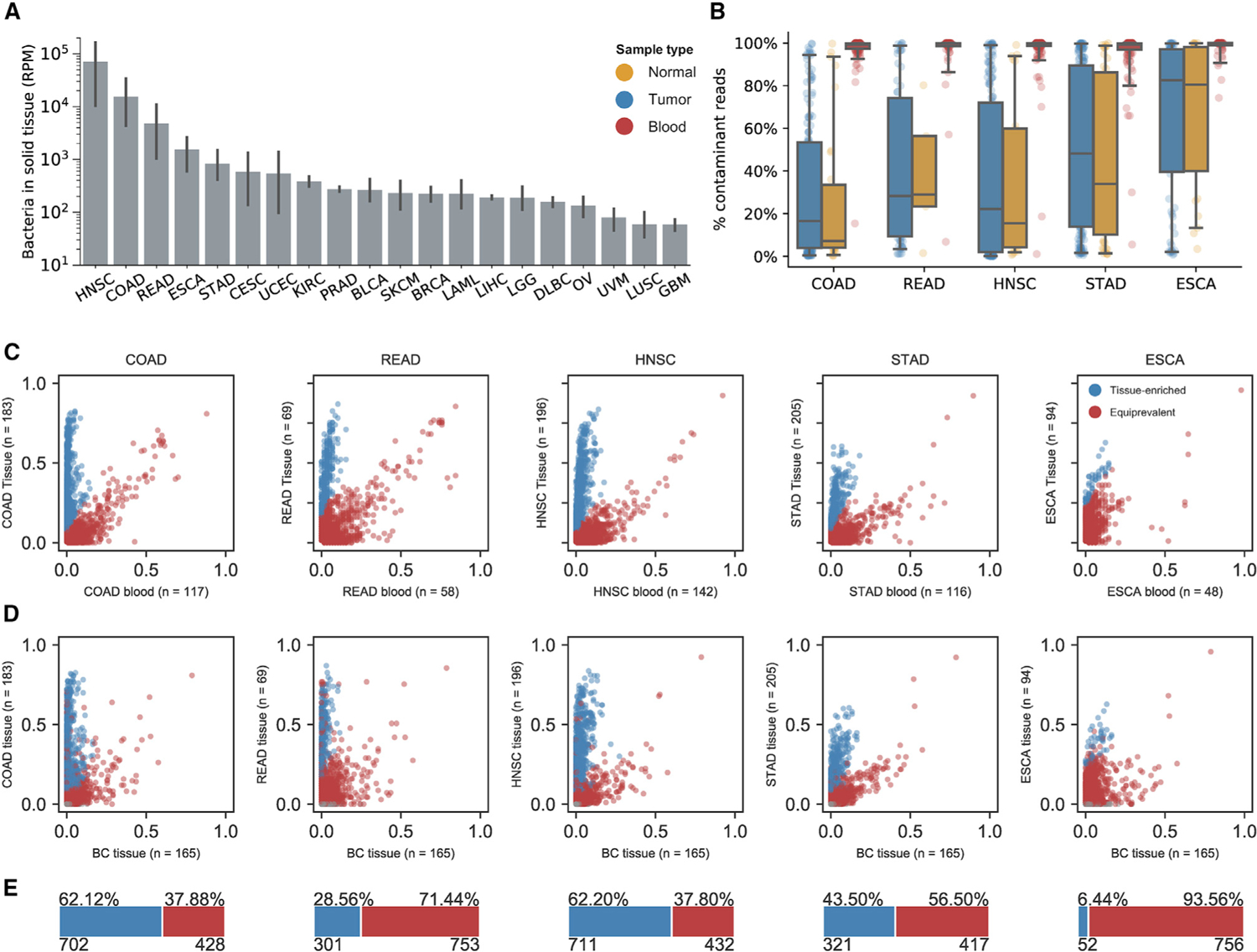
Studying the microbial composition of internal organs and their associations with disease remains challenging due to the difficulty of acquiring clinical biopsies. We designed a statistical model to analyze the prevalence of species across sample types from The Cancer Genome Atlas (TCGA), revealing that species equiprevalent across sample types are predominantly contaminants, bearing unique signatures from each TCGA-designated sequencing center. Removing such species mitigated batch effects and isolated the tissue-resident microbiome, which was validated by original matched TCGA samples. Gene copies and nucleotide variants can further distinguish mixed-evidence species. We, thus, present The Cancer Microbiome Atlas (TCMA), a collection of curated, decontaminated microbial compositions of oropharyngeal, esophageal, gastrointestinal, and colorectal tissues. This led to the discovery of prognostic species and blood signatures of mucosal barrier injuries and enabled systematic matched microbe-host multi-omic analyses, which will help guide future studies of the microbiome's role in human health and disease.
Keywords: colorectal cancer; contamination; host-microbe interactions; human microbiome; multi-omics; pan-cancer; the cancer genome atlas.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
