

Cross-Comparison of Human iPSC Motor Neuron Models of Familial and Sporadic ALS Reveals Early and Convergent Transcriptomic Disease Signatures
- PMID: 33382996
- PMCID: PMC7897311
- DOI: 10.1016/j.cels.2020.10.010
Cross-Comparison of Human iPSC Motor Neuron Models of Familial and Sporadic ALS Reveals Early and Convergent Transcriptomic Disease Signatures
Abstract
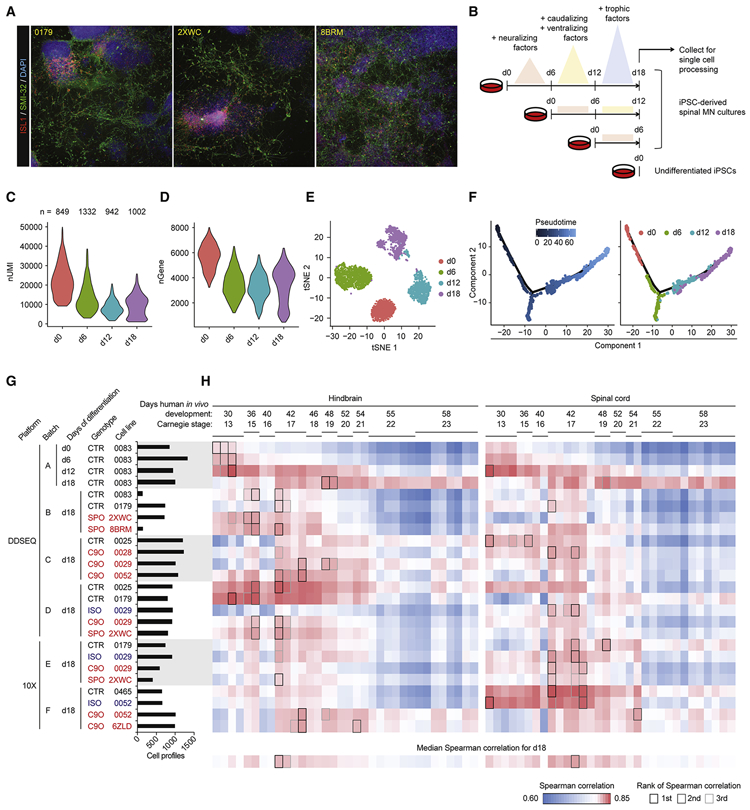
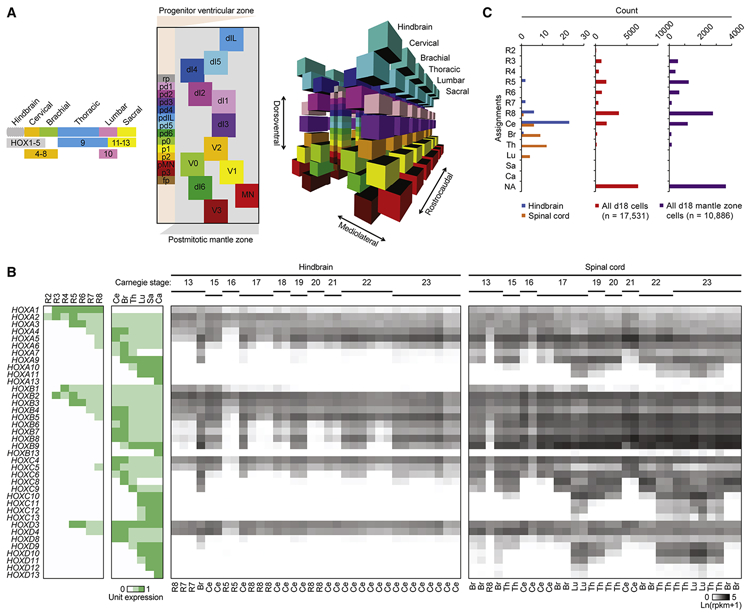
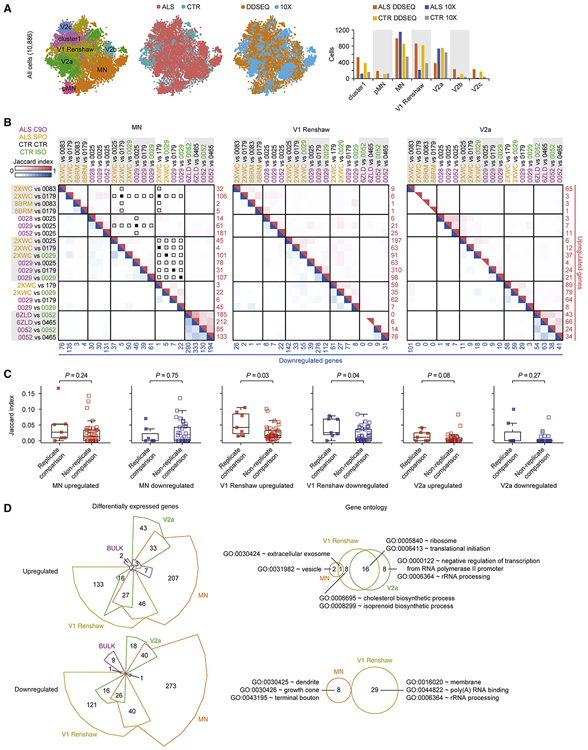
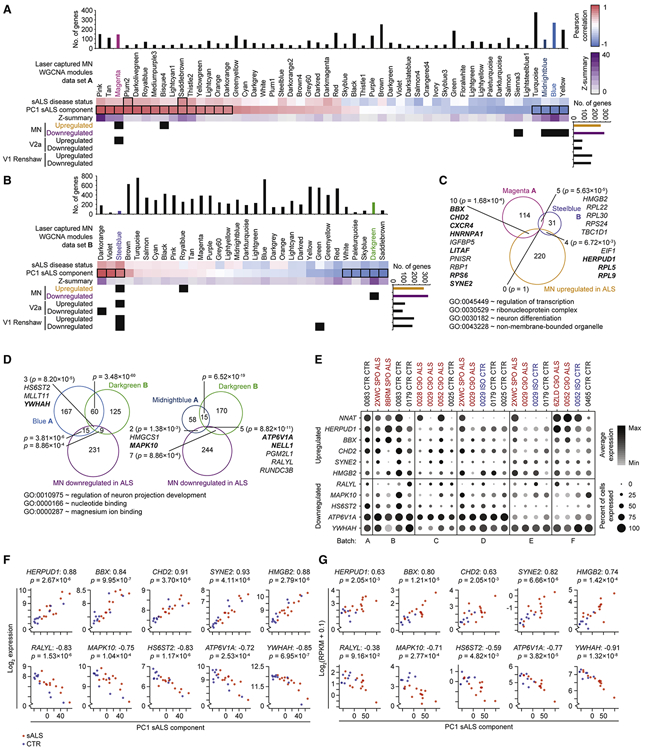
Induced pluripotent stem cell (iPSC)-derived neural cultures from amyotrophic lateral sclerosis (ALS) patients can model disease phenotypes. However, heterogeneity arising from genetic and experimental variability limits their utility, impacting reproducibility and the ability to track cellular origins of pathogenesis. Here, we present methodologies using single-cell RNA sequencing (scRNA-seq) analysis to address these limitations. By repeatedly differentiating and applying scRNA-seq to motor neurons (MNs) from healthy, familial ALS, sporadic ALS, and genome-edited iPSC lines across multiple patients, batches, and platforms, we account for genetic and experimental variability toward identifying unified and reproducible ALS signatures. Combining HOX and developmental gene expression with global clustering, we anatomically classified cells into rostrocaudal, progenitor, and postmitotic identities. By relaxing statistical thresholds, we discovered genes in iPSC-MNs that were concordantly dysregulated in postmortem MNs and yielded predictive ALS markers in other human and mouse models. Our approach thus revealed early, convergent, and MN-resolved signatures of ALS.
Keywords: ALS; ELAVL3; iPSC; single-cell RNA-seq.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures

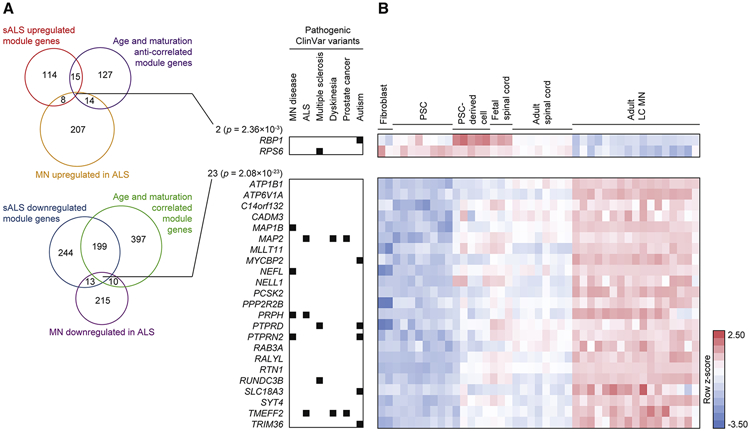
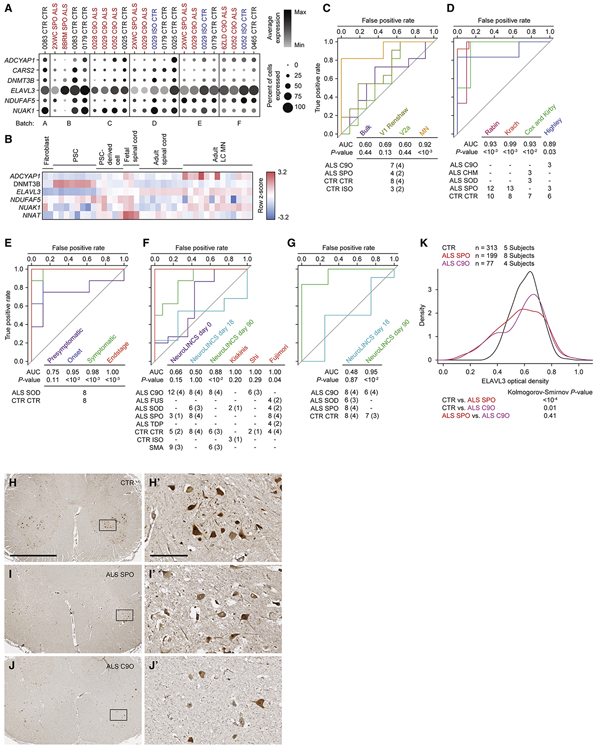
Similar articles
-
Coactivation of GSK3β and IGF-1 Attenuates Amyotrophic Lateral Sclerosis Nerve Fiber Cytopathies in SOD1 Mutant Patient-Derived Motor Neurons.Cells. 2021 Oct 16;10(10):2773. doi: 10.3390/cells10102773. Cells. 2021. PMID: 34685754 Free PMC article.
-
Genetic factors affecting survival in Japanese patients with sporadic amyotrophic lateral sclerosis: a genome-wide association study and verification in iPSC-derived motor neurons from patients.J Neurol Neurosurg Psychiatry. 2023 Oct;94(10):816-824. doi: 10.1136/jnnp-2022-330851. Epub 2023 May 4. J Neurol Neurosurg Psychiatry. 2023. PMID: 37142397
-
Muscle cells of sporadic amyotrophic lateral sclerosis patients secrete neurotoxic vesicles.J Cachexia Sarcopenia Muscle. 2022 Apr;13(2):1385-1402. doi: 10.1002/jcsm.12945. Epub 2022 Feb 22. J Cachexia Sarcopenia Muscle. 2022. PMID: 35194965 Free PMC article.
-
Reverse engineering human neurodegenerative disease using pluripotent stem cell technology.Brain Res. 2016 May 1;1638(Pt A):30-41. doi: 10.1016/j.brainres.2015.09.023. Epub 2015 Sep 28. Brain Res. 2016. PMID: 26423934 Free PMC article. Review.
-
Recent insights from human induced pluripotent stem cell models into the role of microglia in amyotrophic lateral sclerosis.Bioessays. 2024 Jul;46(7):e2400054. doi: 10.1002/bies.202400054. Epub 2024 May 7. Bioessays. 2024. PMID: 38713169 Review.
Cited by
-
ALS molecular subtypes are a combination of cellular and pathological features learned by deep multiomics classifiers.Cell Rep. 2025 Mar 25;44(3):115402. doi: 10.1016/j.celrep.2025.115402. Epub 2025 Mar 10. Cell Rep. 2025. PMID: 40067829 Free PMC article.
-
Functional genomics and the future of iPSCs in disease modeling.Stem Cell Reports. 2022 May 10;17(5):1033-1047. doi: 10.1016/j.stemcr.2022.03.019. Epub 2022 Apr 28. Stem Cell Reports. 2022. PMID: 35487213 Free PMC article. Review.
-
TDP-43-stratified single-cell proteomics of postmortem human spinal motor neurons reveals protein dynamics in amyotrophic lateral sclerosis.Cell Rep. 2024 Jan 23;43(1):113636. doi: 10.1016/j.celrep.2023.113636. Epub 2024 Jan 5. Cell Rep. 2024. PMID: 38183652 Free PMC article.
-
Rapid and Robust Multi-Phenotypic Assay System for ALS Using Human iPS Cells with Mutations in Causative Genes.Int J Mol Sci. 2023 Apr 10;24(8):6987. doi: 10.3390/ijms24086987. Int J Mol Sci. 2023. PMID: 37108151 Free PMC article.
-
Exploring Motor Neuron Diseases Using iPSC Platforms.Stem Cells. 2022 Mar 3;40(1):2-13. doi: 10.1093/stmcls/sxab006. Stem Cells. 2022. PMID: 35511862 Free PMC article.
References
-
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp 2008, P10008.
-
- Büttner M, Miao Z, Wolf FA, Teichmann SA, and Theis FJ (2019). A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
