

Allele-specific expression: applications in cancer and technical considerations
- PMID: 33383480
- PMCID: PMC7985293
- DOI: 10.1016/j.gde.2020.10.007
Allele-specific expression: applications in cancer and technical considerations
Abstract
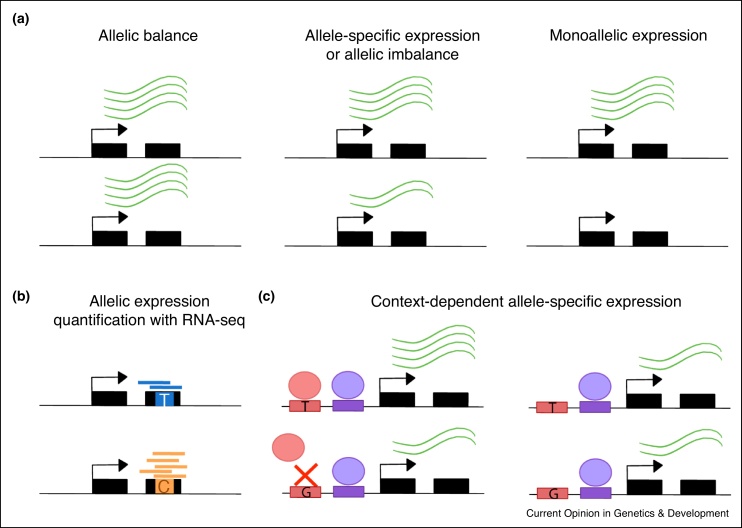
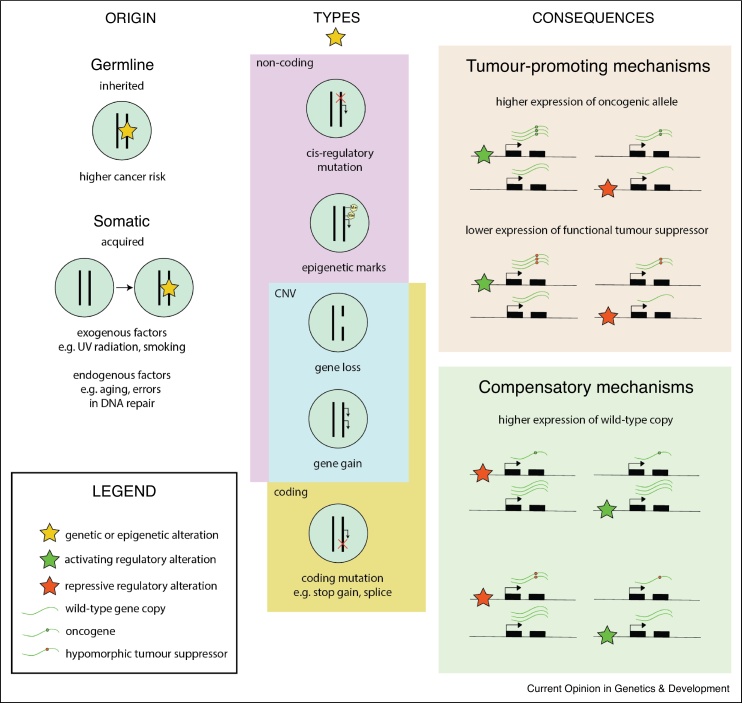
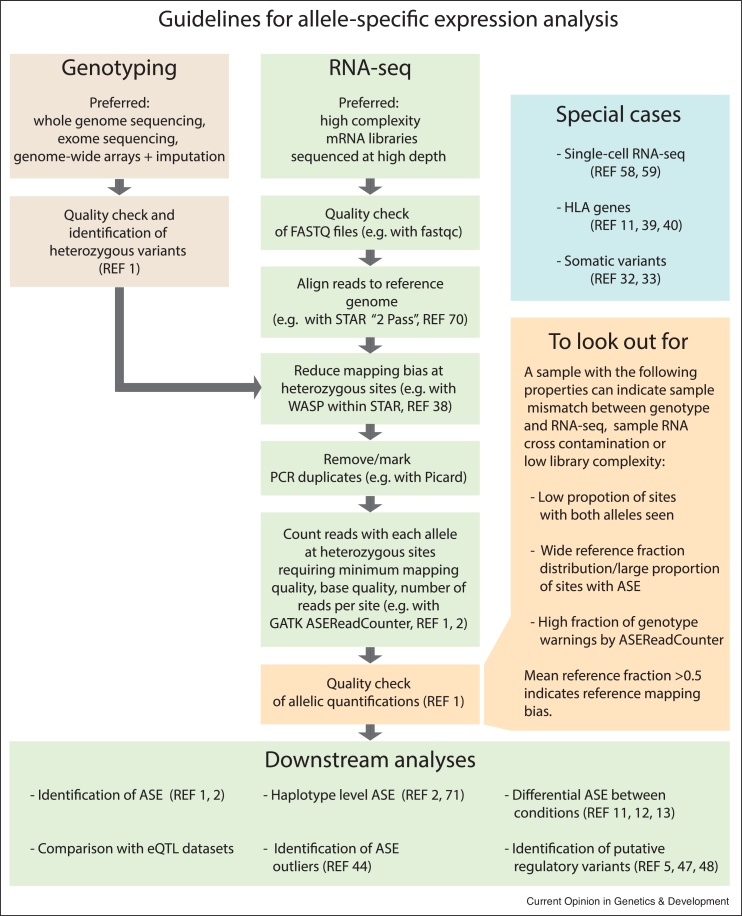
Allele-specific gene expression can influence disease traits. Non-coding germline genetic variants that alter regulatory elements can cause allele-specific gene expression and contribute to cancer susceptibility. In tumors, both somatic copy number alterations and somatic single nucleotide variants have been shown to lead to allele-specific expression of genes, many of which are considered drivers of tumor growth. Here, we review recent studies revealing the pervasive presence of this phenomenon in cancer susceptibility and progression. Furthermore, we underscore the importance of careful experimental design and computational analysis for accurate allelic expression quantification and avoidance of false positives. Finally, we discuss additional methodological challenges encountered in cancer studies and in the burgeoning field of single-cell transcriptomics.
Copyright © 2021. Published by Elsevier Ltd.
Figures
References
-
- Castel S.E., GTEx Consortium, Aguet F., Mohammadi P., Ardlie K.G., Lappalainen T. A vast resource of allelic expression data spanning human tissues. Genome Biol. 2020;21 - PMC - PubMed
-
Using a large RNA-seq resource of 54 tissues from 838 individuals, the authors identify allele-specific expression across 15,253 samples using exemplary methods. They identify ASE at both the SNP level and haplotype level, and present a new tool to estimate effect sizes of cis-regulatory variants.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
