

Genotype-Phenotype Correlations in Central Precocious Puberty Caused by MKRN3 Mutations
- PMID: 33383582
- PMCID: PMC7993586
- DOI: 10.1210/clinem/dgaa955
Genotype-Phenotype Correlations in Central Precocious Puberty Caused by MKRN3 Mutations
Erratum in
-
Corrigendum to: "Genotype-Phenotype Correlations in Central Precocious Puberty Caused by MKRN3 Mutations".J Clin Endocrinol Metab. 2021 Oct 21;106(11):e4794. doi: 10.1210/clinem/dgab394. J Clin Endocrinol Metab. 2021. PMID: 34173650 Free PMC article. No abstract available.
Abstract
Context: Loss-of-function mutations of makorin RING finger protein 3 (MKRN3) are the most common monogenic cause of familial central precocious puberty (CPP).
Objective: To describe the clinical and hormonal features of a large cohort of patients with CPP due to MKRN3 mutations and compare the characteristics of different types of genetic defects.
Methods: Multiethnic cohort of 716 patients with familial or idiopathic CPP screened for MKRN3 mutations using Sanger sequencing. A group of 156 Brazilian girls with idiopathic CPP (ICPP) was used as control group.
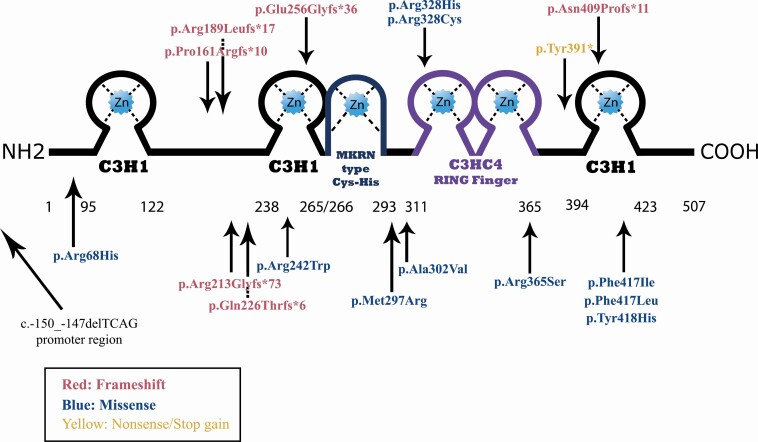
Results: Seventy-one patients (45 girls and 26 boys from 36 families) had 18 different loss-of-function MKRN3 mutations. Eight mutations were classified as severe (70% of patients). Among the 71 patients, first pubertal signs occurred at 6.2 ± 1.2 years in girls and 7.1 ± 1.5 years in boys. Girls with MKRN3 mutations had a shorter delay between puberty onset and first evaluation and higher follicle-stimulating hormone levels than ICPP. Patients with severe MKRN3 mutations had a greater bone age advancement than patients with missense mutations (2.3 ± 1.6 vs 1.6 ± 1.4 years, P = .048), and had higher basal luteinizing hormone levels (2.2 ± 1.8 vs 1.1 ± 1.1 UI/L, P = .018) at the time of presentation. Computational protein modeling revealed that 60% of the missense mutations were predicted to cause protein destabilization.
Conclusion: Inherited premature activation of the reproductive axis caused by loss-of-function mutations of MKRN3 is clinically indistinct from ICPP. However, the type of genetic defect may affect bone age maturation and gonadotropin levels.
Keywords: MKRN3; MKRN3 phenotype; genetic of puberty; precocious puberty.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Jong MT, Gray TA, Ji Y, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet. 1999;8(5):783-793. - PubMed
-
- Nicholls RD, Saitoh S, Horsthemke B. Imprinting in Prader-Willi and Angelman syndromes. Trends Genet. 1998;14(5):194-200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
