

Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency
- PMID: 33385162
- PMCID: PMC7751686
- DOI: 10.1093/narcan/zcaa038
Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency
Abstract
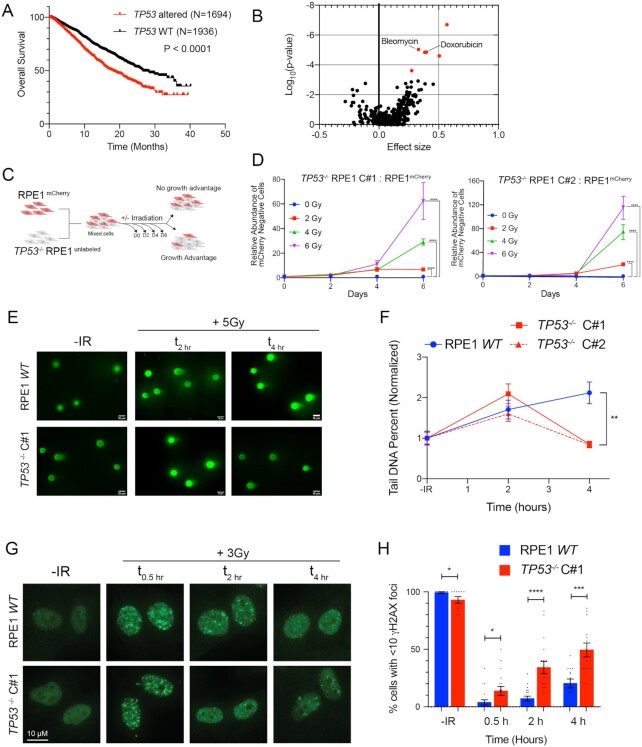
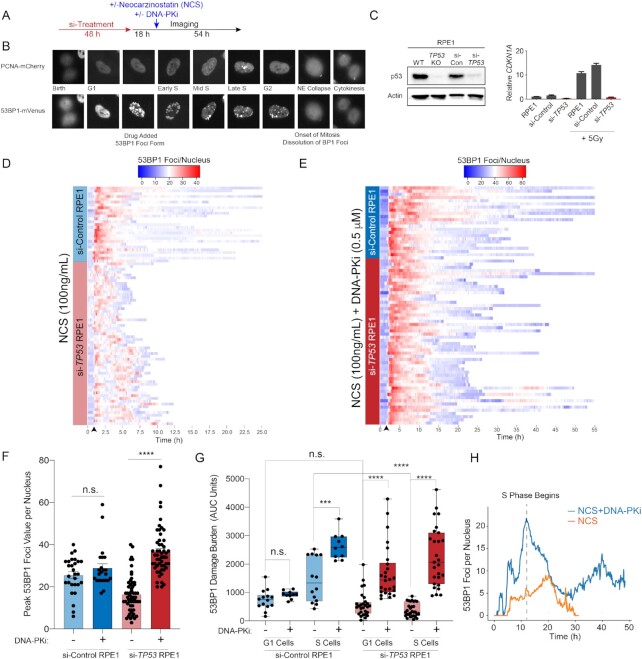
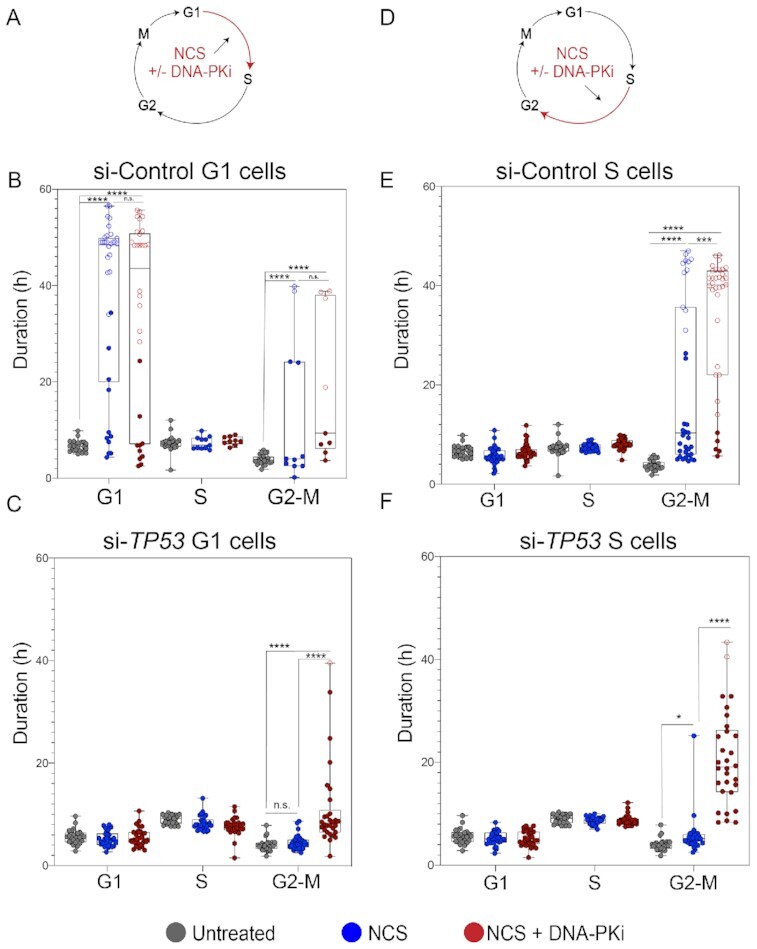
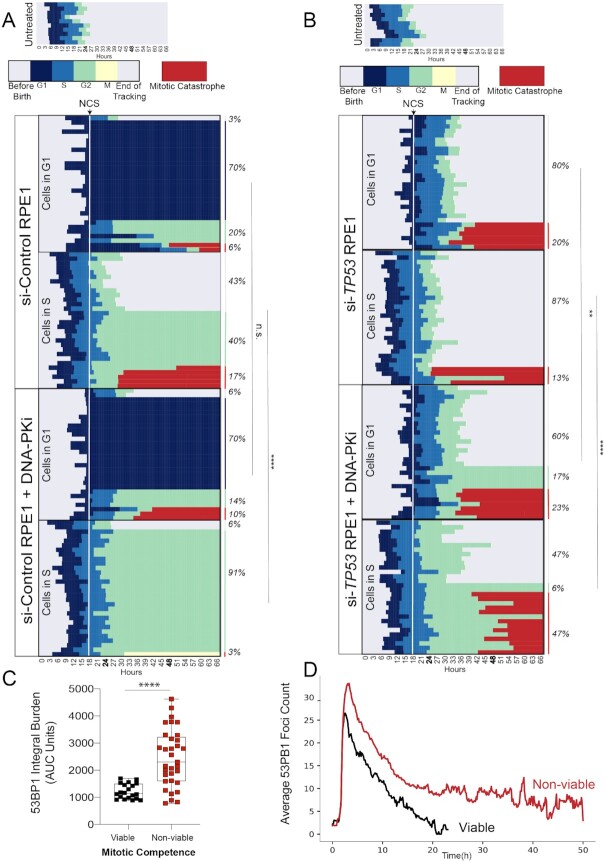
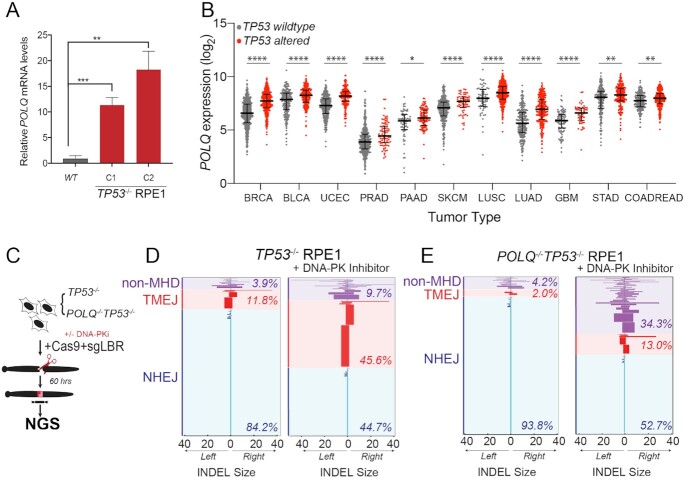
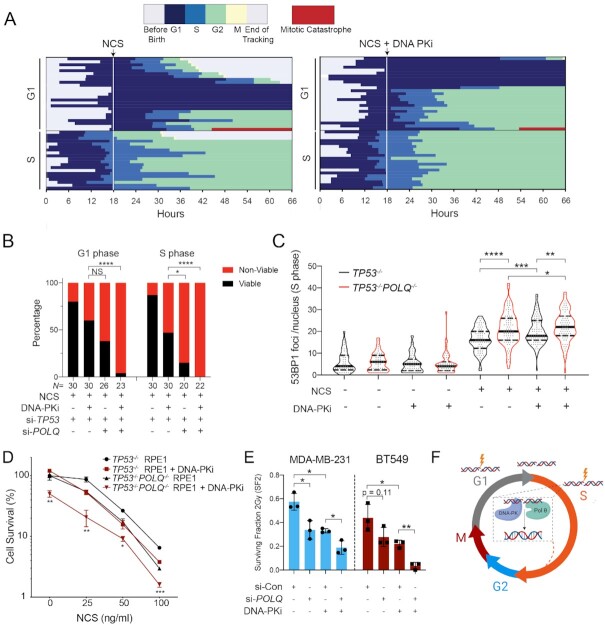
TP53 deficiency in cancer is associated with poor patient outcomes and resistance to DNA damaging therapies. However, the mechanisms underlying treatment resistance in p53-deficient cells remain poorly characterized. Using live cell imaging of DNA double-strand breaks (DSBs) and cell cycle state transitions, we show that p53-deficient cells exhibit accelerated repair of radiomimetic-induced DSBs arising in S phase. Low-dose DNA-dependent protein kinase (DNA-PK) inhibition increases the S-phase DSB burden in p53-deficient cells, resulting in elevated rates of mitotic catastrophe. However, a subset of p53-deficient cells exhibits intrinsic resistance to radiomimetic-induced DSBs despite DNA-PK inhibition. We show that p53-deficient cells under DNA-PK inhibition utilize DNA polymerase theta (Pol θ)-mediated end joining repair to promote their viability in response to therapy-induced DSBs. Pol θ inhibition selectively increases S-phase DSB burden after radiomimetic therapy and promotes prolonged G2 arrest. Dual inhibition of DNA-PK and Pol θ restores radiation sensitivity in p53-deficient cells as well as in p53-mutant breast cancer cell lines. Thus, combination targeting of DNA-PK- and Pol θ-dependent end joining repair represents a promising strategy for overcoming resistance to DNA damaging therapies in p53-deficient cancers.
© The Author(s) 2020. Published by Oxford University Press on behalf of NAR Cancer.
Figures
References
-
- Nigro J.M., Baker S.J., Preisinger A.C., Jessup J.M., Hostetter R., Cleary K., Bigner S.H., Davidson N., Baylin S., Devilee P. et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989; 342:705–708. - PubMed
-
- Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019; 20:199–210. - PubMed
-
- Miller L.D., Smeds J., George J., Vega V.B., Vergara L., Ploner A., Pawitan Y., Hall P., Klaar S., Liu E.T. et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl Acad. Sci. U.S.A. 2005; 102:13550–13555. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
