

Sensitive universal detection of blood parasites by selective pathogen-DNA enrichment and deep amplicon sequencing
- PMID: 33388088
- PMCID: PMC7778815
- DOI: 10.1186/s40168-020-00939-1
Sensitive universal detection of blood parasites by selective pathogen-DNA enrichment and deep amplicon sequencing
Abstract
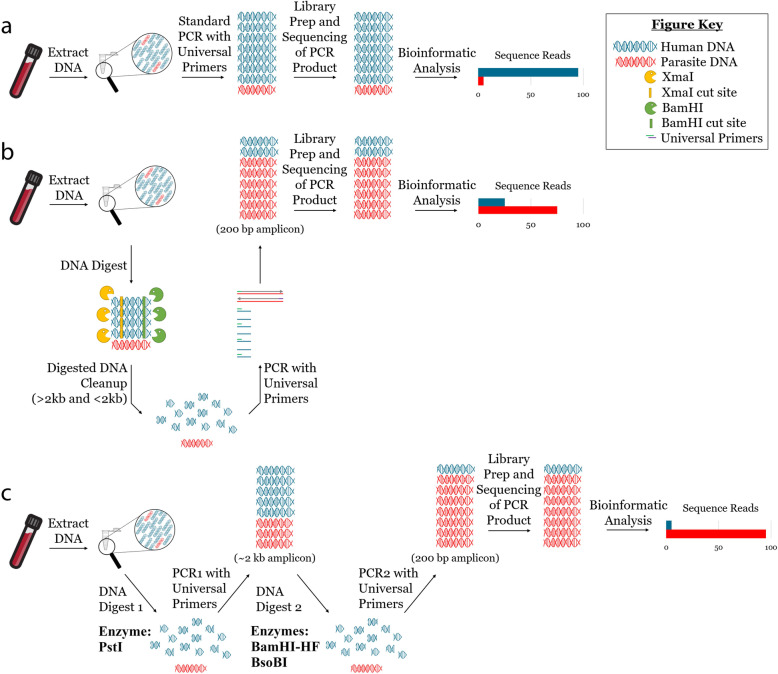
Background: Targeted amplicon deep sequencing (TADS) has enabled characterization of diverse bacterial communities, yet the application of TADS to communities of parasites has been relatively slow to advance. The greatest obstacle to this has been the genetic diversity of parasitic agents, which include helminths, protozoa, arthropods, and some acanthocephalans. Meanwhile, universal amplification of conserved loci from all parasites without amplifying host DNA has proven challenging. Pan-eukaryotic PCRs preferentially amplify the more abundant host DNA, obscuring parasite-derived reads following TADS. Flaherty et al. (2018) described a pan-parasitic TADS method involving amplification of eukaryotic 18S rDNA regions possessing restriction sites only in vertebrates. Using this method, host DNA in total DNA extracts could be selectively digested prior to PCR using restriction enzymes, thereby increasing the number of parasite-derived reads obtained following NGS. This approach showed promise though was only as sensitive as conventional PCR.
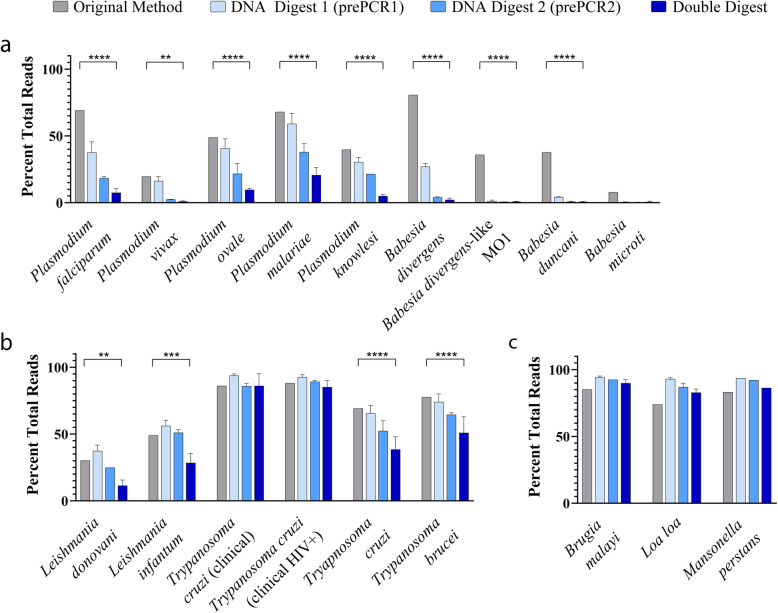
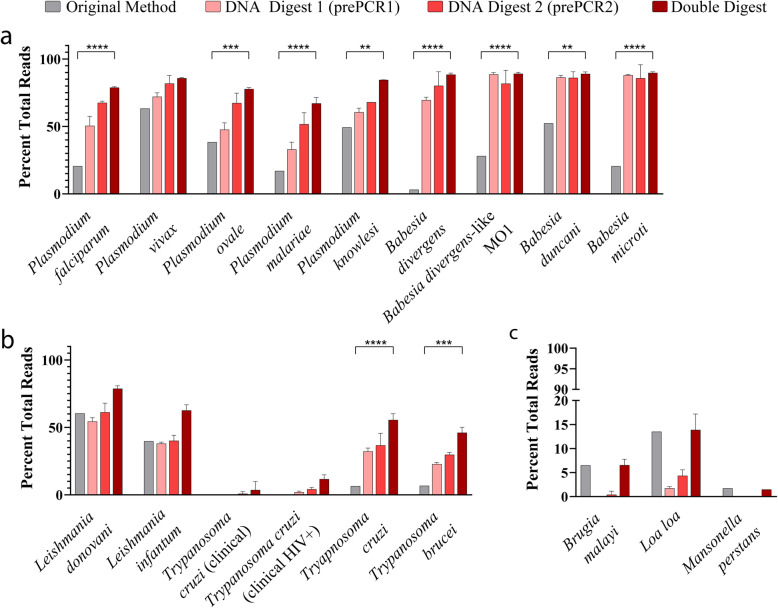
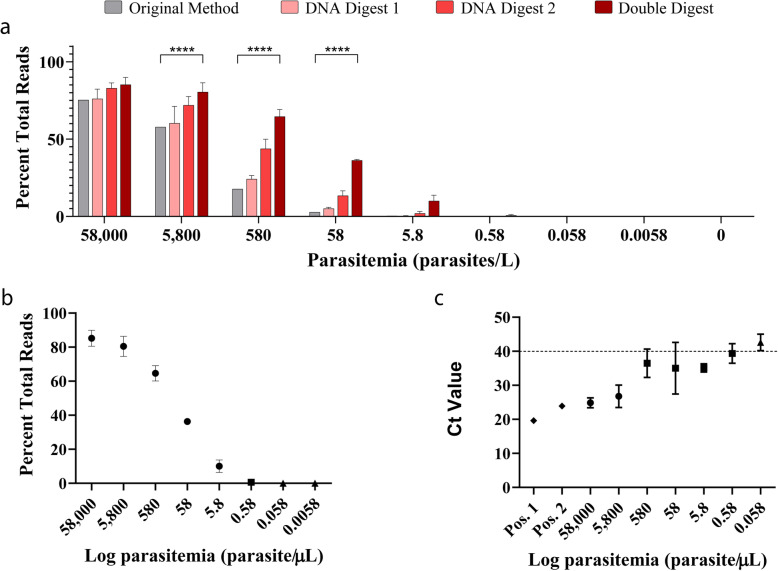
Results: Here, we expand on this work by designing a second set of pan-eukaryotic primers flanking the priming sites already described, enabling nested PCR amplification of the established 18S rDNA target. This nested approach facilitated introduction of a second restriction digestion between the first and second PCR, reducing the proportional mass of amplifiable host-derived DNA while increasing the number of PCR amplification cycles. We applied this method to blood specimens containing Babesia, Plasmodium, various kinetoplastids, and filarial nematodes and confirmed its limit of detection (LOD) to be approximately 10-fold lower than previously described, falling within the range of most qPCR methods.
Conclusions: The assay detects and differentiates the major malaria parasites of humans, along with several other clinically important blood parasites. This represents an important step towards a TADS-based universal parasite diagnostic (UPDx) test with a sufficient LOD for routine applications. Video Abstract.
Keywords: Amplicon sequencing; Blood microbiota; Molecular diagnosis; Molecular parasitology; Parasite biodiversity; Parasite detection.
Conflict of interest statement
The authors of this manuscript have no competing interests to disclose.
Figures
Similar articles
-
Restriction enzyme digestion of host DNA enhances universal detection of parasitic pathogens in blood via targeted amplicon deep sequencing.Microbiome. 2018 Sep 17;6(1):164. doi: 10.1186/s40168-018-0540-2. Microbiome. 2018. PMID: 30223888 Free PMC article.
-
Simultaneous targeted amplicon deep sequencing and library preparation for a time and cost-effective universal parasite diagnostic sequencing approach.Parasitol Res. 2023 Dec;122(12):3243-3256. doi: 10.1007/s00436-023-07991-4. Epub 2023 Nov 9. Parasitol Res. 2023. PMID: 37940706 Free PMC article.
-
Assessing an Adaptation of the Universal Parasite Diagnostic Assay for Bloodborne Parasites in a US State Public Health Laboratory.Am J Trop Med Hyg. 2021 Nov 9;106(2):671-677. doi: 10.4269/ajtmh.21-0707. Am J Trop Med Hyg. 2021. PMID: 34749306 Free PMC article.
-
Digital PCR: a new technology for diagnosis of parasitic infections.Clin Microbiol Infect. 2019 Dec;25(12):1510-1516. doi: 10.1016/j.cmi.2019.06.009. Epub 2019 Jun 18. Clin Microbiol Infect. 2019. PMID: 31226445 Review.
-
Molecular diagnosis in clinical parasitology: when and why?Exp Biol Med (Maywood). 2014 Nov;239(11):1443-60. doi: 10.1177/1535370214523880. Epub 2014 Mar 25. Exp Biol Med (Maywood). 2014. PMID: 24668556 Review.
Cited by
-
A Recent Advance in the Diagnosis, Treatment, and Vaccine Development for Human Schistosomiasis.Trop Med Infect Dis. 2024 Oct 15;9(10):243. doi: 10.3390/tropicalmed9100243. Trop Med Infect Dis. 2024. PMID: 39453270 Free PMC article. Review.
-
Molecular diagnosis of infectious parasites in the post-COVID-19 era.Trop Parasitol. 2021 Jan-Jun;11(1):3-10. doi: 10.4103/tp.tp_12_21. Epub 2021 May 14. Trop Parasitol. 2021. PMID: 34195053 Free PMC article.
-
A Generalized Bayesian Stochastic Block Model for Microbiome Community Detection.Stat Med. 2025 Feb 10;44(3-4):e10291. doi: 10.1002/sim.10291. Stat Med. 2025. PMID: 39853798 Free PMC article.
-
The Troublesome Ticks Research Protocol: Developing a Comprehensive, Multidiscipline Research Plan for Investigating Human Tick-Associated Disease in Australia.Pathogens. 2022 Nov 3;11(11):1290. doi: 10.3390/pathogens11111290. Pathogens. 2022. PMID: 36365042 Free PMC article.
-
Nested qPCR assay to detect Babesia duncani infection in hamsters and humans.Parasitol Res. 2022 Dec;121(12):3603-3610. doi: 10.1007/s00436-022-07685-3. Epub 2022 Oct 4. Parasitol Res. 2022. PMID: 36192649
References
-
- Talundzic E, Ravishankar S, Kelley J, Patel D, Plucinski M, Schmedes S, Ljolje D, Clemons B, Madison-Antenucci S, Arguin PM, et al. Next-generation sequencing and bioinformatics protocol for malaria drug resistance marker surveillance. Antimicrob Agents Chemother. 2018;62(4):e02474–e02417. doi: 10.1128/AAC.02474-17. - DOI - PMC - PubMed
-
- Van Borm S, Belak S, Freimanis G, Fusaro A, Granberg F, Hoper D, King DP, Monne I, Orton R, Rosseel T. Next-generation sequencing in veterinary medicine: how can the massive amount of information arising from high-throughput technologies improve diagnosis, control, and management of infectious diseases? Methods Mol Biol. 2015;1247:415–436. doi: 10.1007/978-1-4939-2004-4_30. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
