

Dynamics of severe acute respiratory syndrome coronavirus 2 genome variants in the feces during convalescence
- PMID: 33388272
- PMCID: PMC7649052
- DOI: 10.1016/j.jgg.2020.10.002
Dynamics of severe acute respiratory syndrome coronavirus 2 genome variants in the feces during convalescence
Abstract
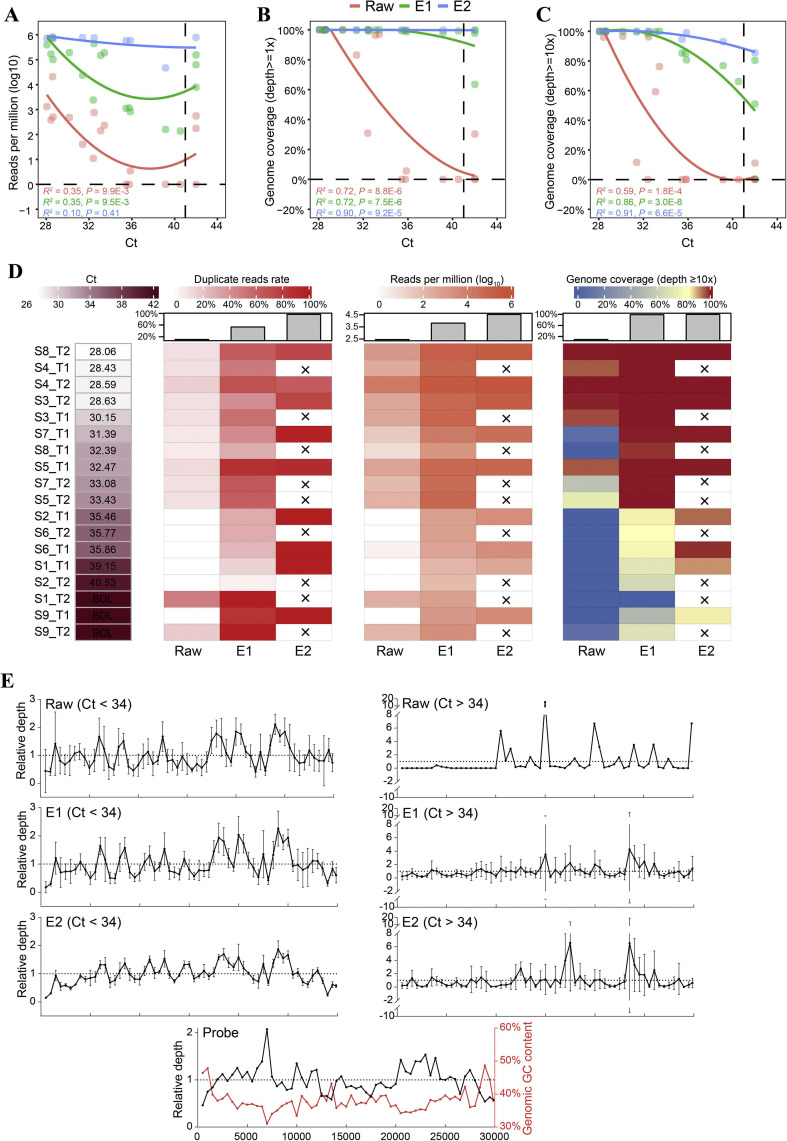
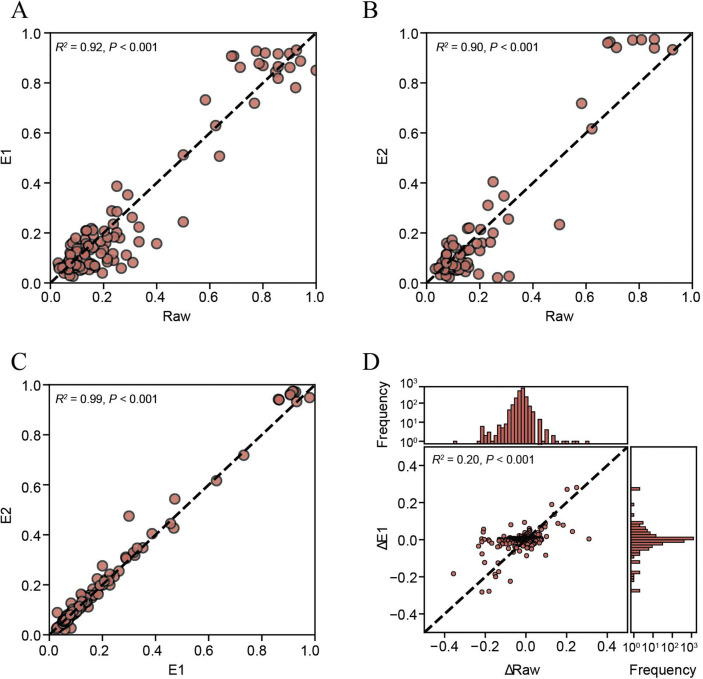
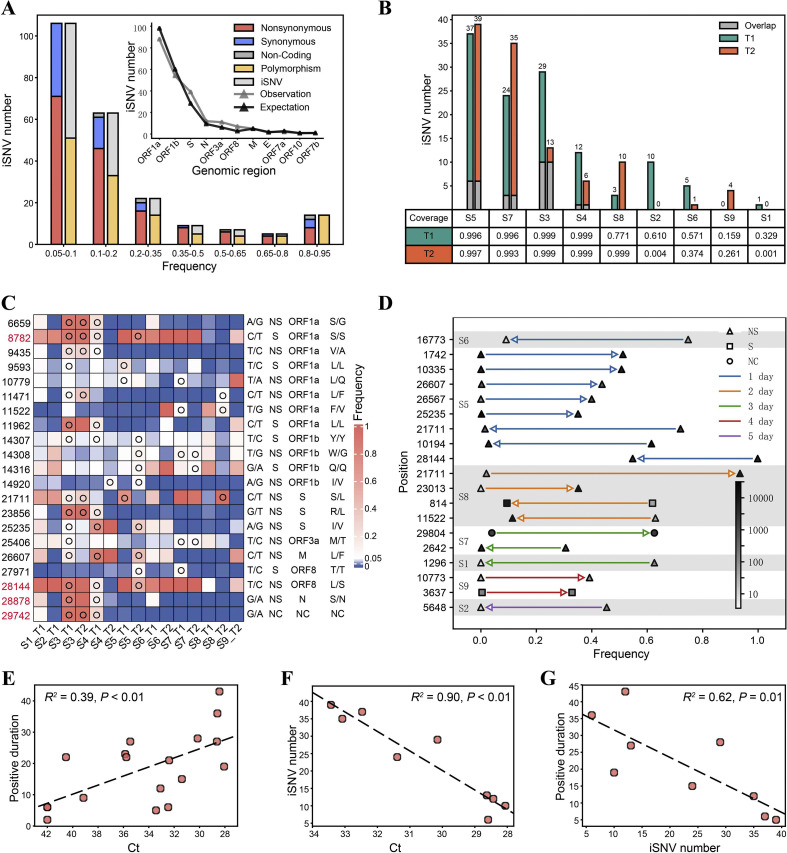
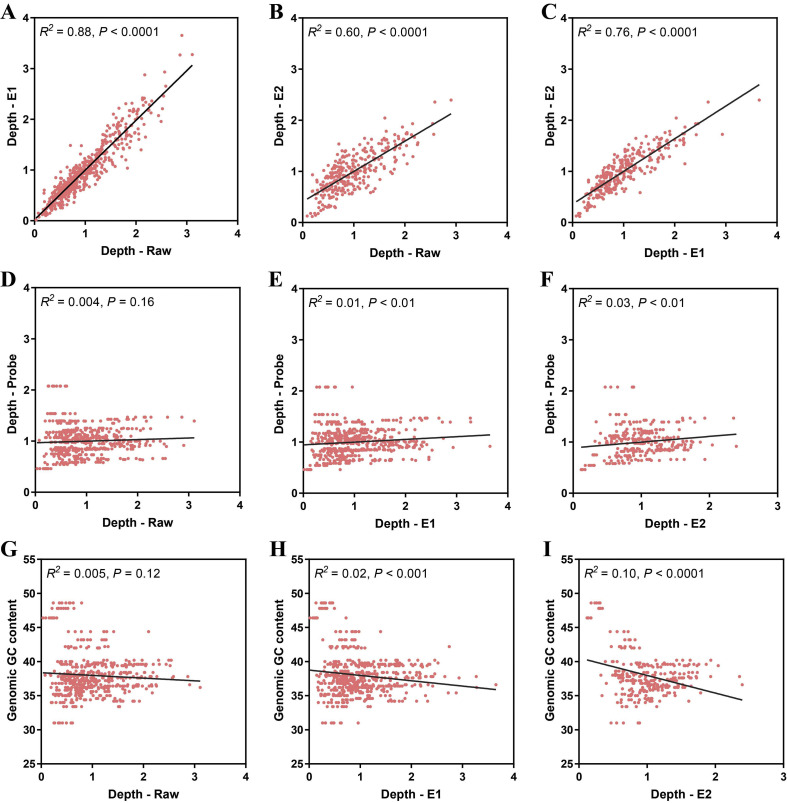
In response to the current coronavirus disease 2019 (COVID-19) pandemic, it is crucial to understand the origin, transmission, and evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which relies on close surveillance of genomic diversity in clinical samples. Although the mutation at the population level had been extensively investigated, how the mutations evolve at the individual level is largely unknown. Eighteen time-series fecal samples were collected from nine patients with COVID-19 during the convalescent phase. The nucleic acids of SARS-CoV-2 were enriched by the hybrid capture method. First, we demonstrated the outstanding performance of the hybrid capture method in detecting intra-host variants. We identified 229 intra-host variants at 182 sites in 18 fecal samples. Among them, nineteen variants presented frequency changes > 0.3 within 1-5 days, reflecting highly dynamic intra-host viral populations. Moreover, the evolution of the viral genome demonstrated that the virus was probably viable in the gastrointestinal tract during the convalescent period. Meanwhile, we also found that the same mutation showed a distinct pattern of frequency changes in different individuals, indicating a strong random drift. In summary, dramatic changes of the SARS-CoV-2 genome were detected in fecal samples during the convalescent period; whether the viral load in feces is sufficient to establish an infection warranted further investigation.
Keywords: Dynamics; Hybrid capture; Intra-host variant; Mutation; SARS-CoV-2.
Copyright © 2020 Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and Genetics Society of China. Published by Elsevier Ltd. All rights reserved.
Figures
References
-
- Farci P., Shimoda A., Coiana A., Diaz G., Peddis G., Melpolder J.C., Strazzera A., Chien D.Y., Munoz S.J., Balestrieri A., Purcell R.H., Alter H.J. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339–344. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
