

Revealing the impact of structural variants in multiple myeloma
- PMID: 33392515
- PMCID: PMC7774871
- DOI: 10.1158/2643-3230.BCD-20-0132
Revealing the impact of structural variants in multiple myeloma
Abstract
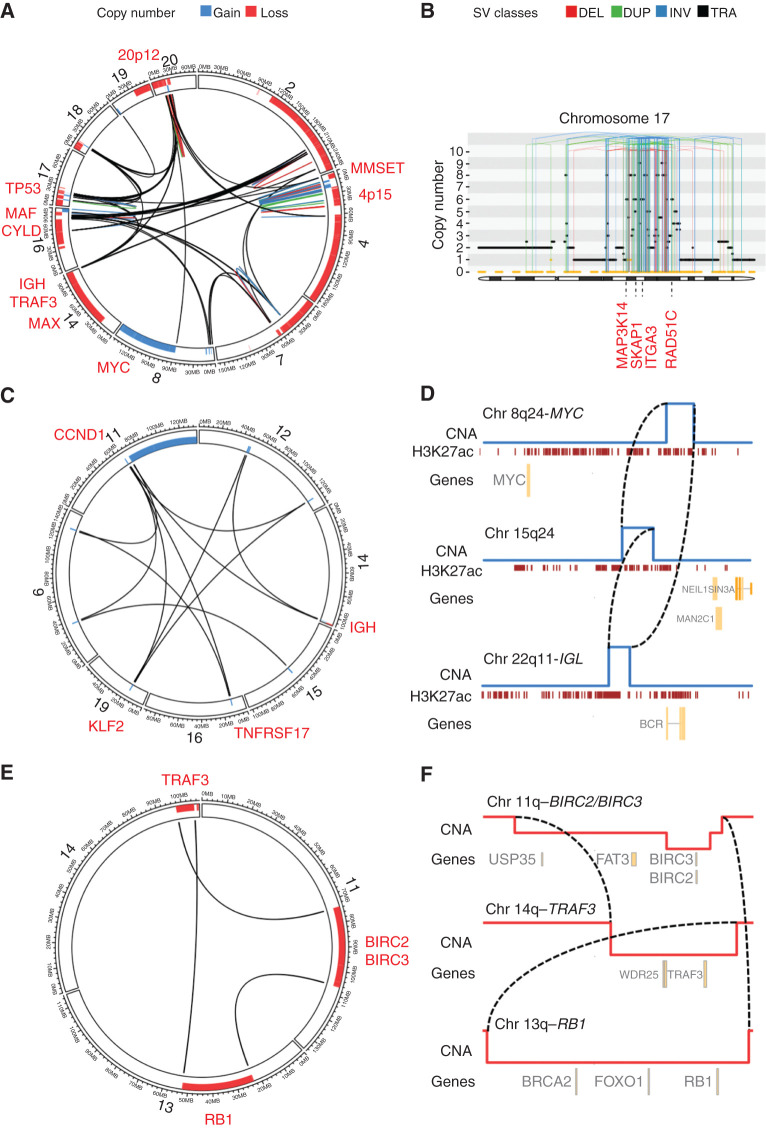
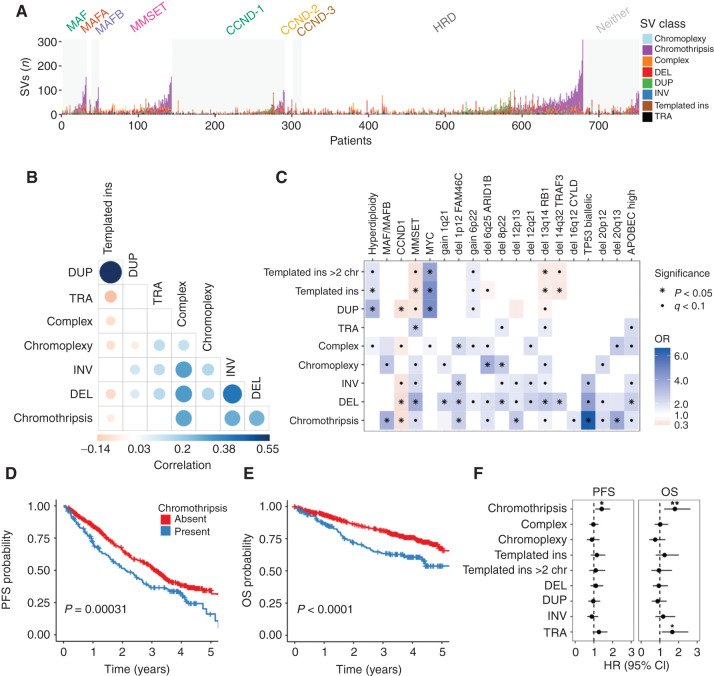
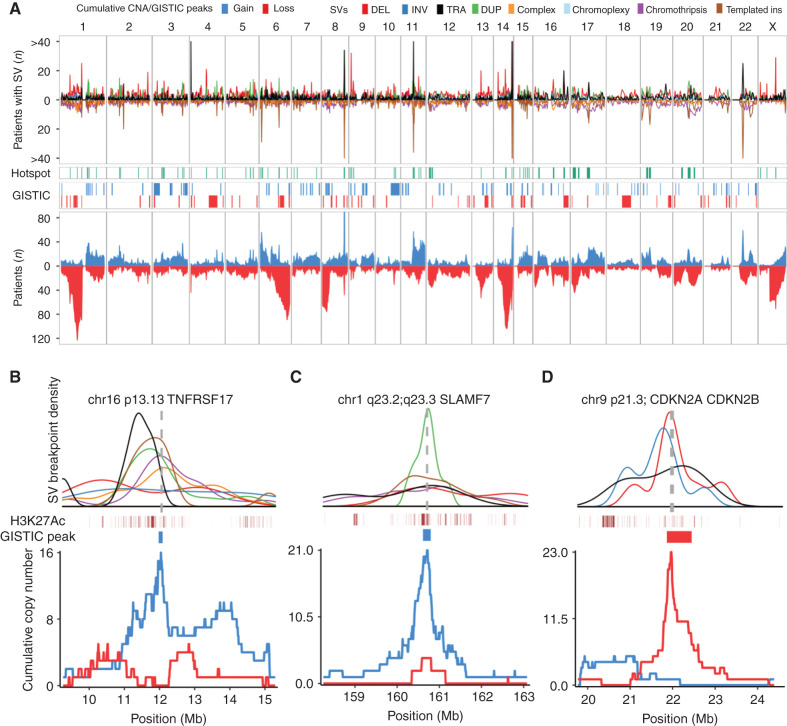
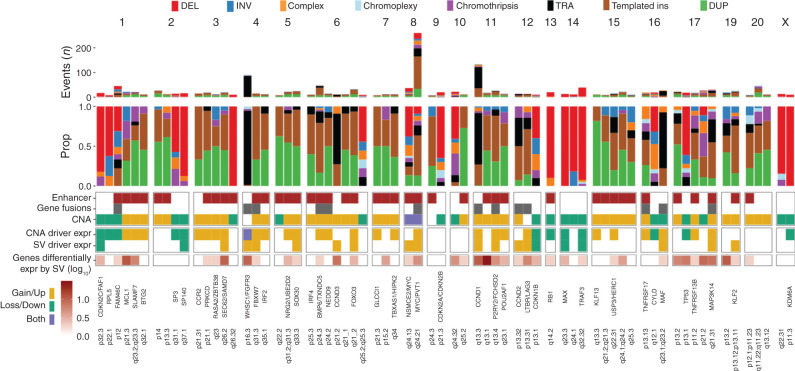
The landscape of structural variants (SVs) in multiple myeloma remains poorly understood. Here, we performed comprehensive analysis of SVs in a large cohort of 752 multiple myeloma patients by low coverage long-insert whole genome sequencing. We identified 68 SV hotspots involving 17 new candidate driver genes, including the therapeutic targets BCMA (TNFRSF17), SLAMF and MCL1. Catastrophic complex rearrangements termed chromothripsis were present in 24% of patients and independently associated with poor clinical outcomes. Templated insertions were the second most frequent complex event (19%), mostly involved in super-enhancer hijacking and activation of oncogenes such as CCND1 and MYC. Importantly, in 31% of patients two or more seemingly independent putative driver events were caused by a single structural event, demonstrating that the complex genomic landscape of multiple myeloma can be acquired through few key events during tumor evolutionary history. Overall, this study reveals the critical role of SVs in multiple myeloma pathogenesis.
Keywords: chromoplexy; chromothripsis; hotspots; multiple myeloma; structural variants; templated insertion.
Conflict of interest statement
Conflict of interest statement No conflict of interests to declare.
Figures



Comment in
-
Promiscuous Structural Variants Drive Myeloma Initiation and Progression.Blood Cancer Discov. 2020 Oct 15;1(3):221-223. doi: 10.1158/2643-3230.BCD-20-0170. eCollection 2020 Nov. Blood Cancer Discov. 2020. PMID: 34661146 Free PMC article.
References
-
- Lee JJ, Park S, Park H, Kim S, Lee J, Lee J, et al. Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma. Cell 2019;177:1842–57. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
