

Acriflavine, a Potent Inhibitor of HIF-1α, Disturbs Glucose Metabolism and Suppresses ATF4-Protective Pathways in Melanoma under Non-Hypoxic Conditions
- PMID: 33396270
- PMCID: PMC7795823
- DOI: 10.3390/cancers13010102
Acriflavine, a Potent Inhibitor of HIF-1α, Disturbs Glucose Metabolism and Suppresses ATF4-Protective Pathways in Melanoma under Non-Hypoxic Conditions
Abstract
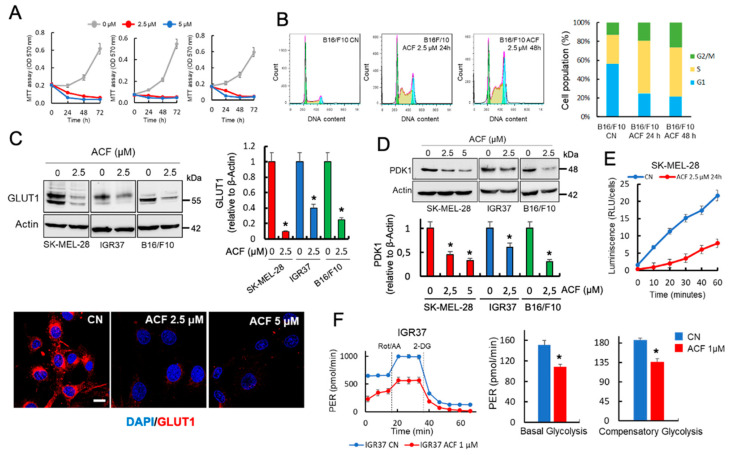
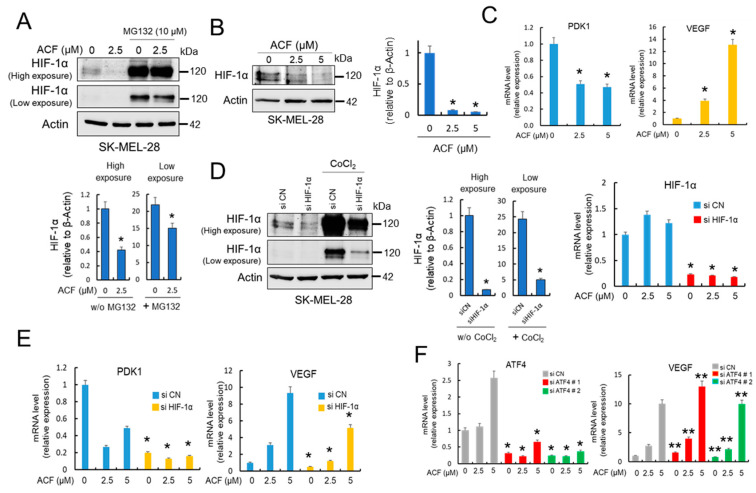
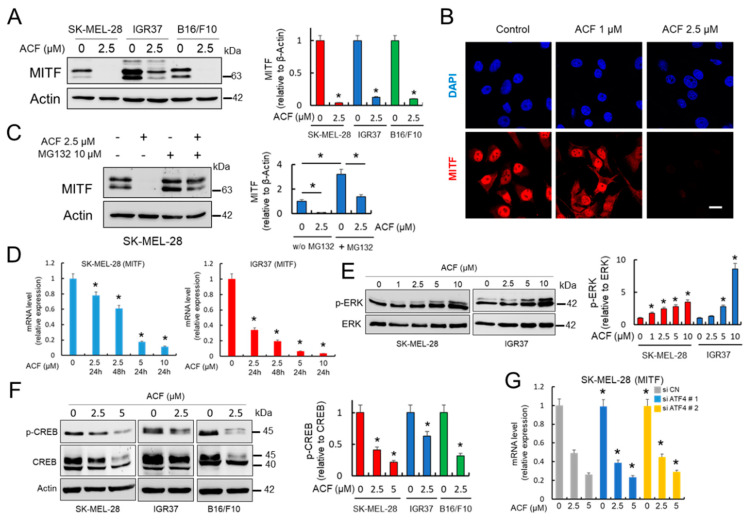
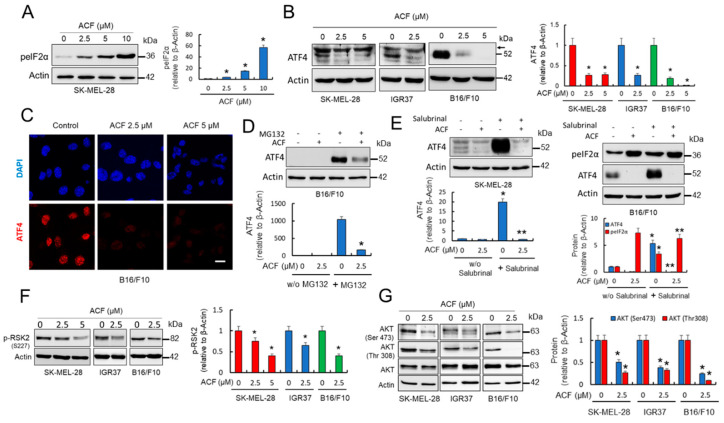
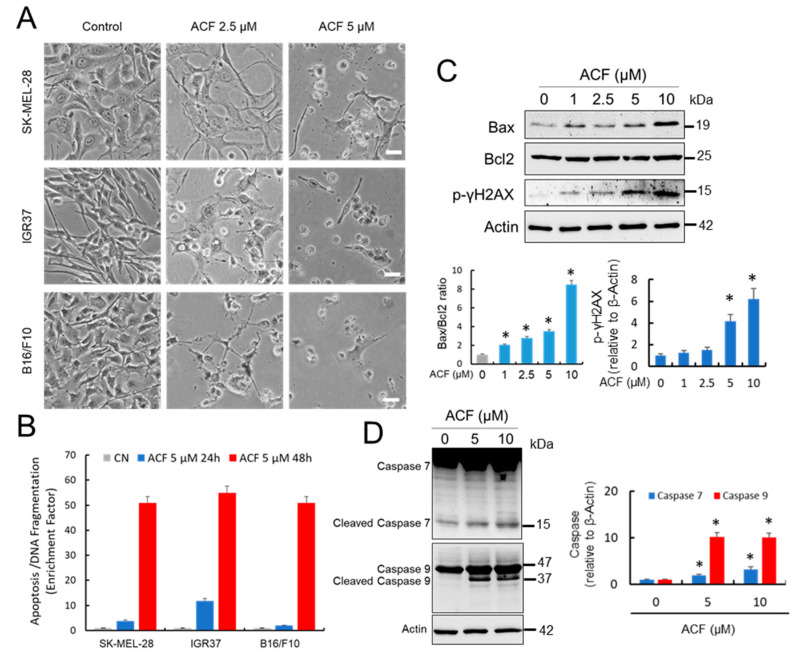
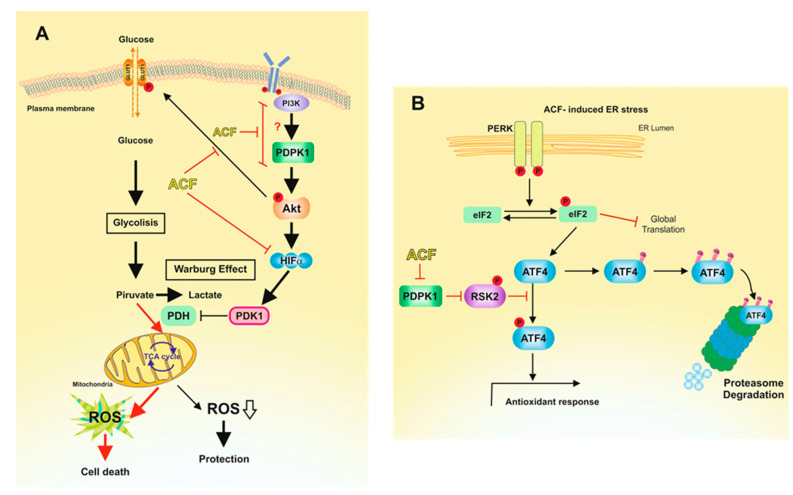
Hypoxia-inducible factor (HIF)-1α is constitutively expressed in melanoma cells under normoxic conditions and its elevated expression correlates with the aggressiveness of melanoma tumors. Here, we used acriflavine, a potent inhibitor of HIF-1α dimerization, as a tool to investigate whether HIF-1α-regulated pathways contribute to the growth of melanoma cells under normoxia. We observed that acriflavine differentially modulated HIF-1α-regulated targets in melanoma under normoxic conditions, although acriflavine treatment resulted in over-expression of vascular endothelial growth factor (VEGF), its action clearly downregulated the expression of pyruvate dehydrogenase kinase 1 (PDK1), a well-known target of HIF-1α. Consequently, downregulation of PDK1 by acrifavine resulted in reduced glucose availability and suppression of the Warburg effect in melanoma cells. In addition, by inhibiting the AKT and RSK2 phosphorylation, acriflavine also avoided protective pathways necessary for survival under conditions of oxidative stress. Interestingly, we show that acriflavine targets activating transcription factor 4 (ATF4) for proteasomal degradation while suppressing the expression of microphthalmia-associated transcription factor (MITF), a master regulator of melanocyte development and a melanoma oncogene. Since acriflavine treatment results in the consistent death of melanoma cells, our results suggest that inhibition of HIF-1α function in melanoma could open new avenues for the treatment of this deadly disease regardless of the hypoxic condition of the tumor.
Keywords: ATF4; HIF-1α; MITF; acriflavine; glucose metabolism; melanoma; oxygen homeostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Powis G., Kirkpatrick L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol. Cancer Ther. 2004;3:647–654. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
