

doi: 10.1186/s13059-020-02229-3.
Reference flow: reducing reference bias using multiple population genomes
Affiliations
- PMID: 33397413
- PMCID: PMC7780692
- DOI: 10.1186/s13059-020-02229-3
Item in Clipboard
Reference flow: reducing reference bias using multiple population genomes
Genome Biol.
.
Abstract
Most sequencing data analyses start by aligning sequencing reads to a linear reference genome, but failure to account for genetic variation leads to reference bias and confounding of results downstream. Other approaches replace the linear reference with structures like graphs that can include genetic variation, incurring major computational overhead. We propose the reference flow alignment method that uses multiple population reference genomes to improve alignment accuracy and reduce reference bias. Compared to the graph aligner vg, reference flow achieves a similar level of accuracy and bias avoidance but with 14% of the memory footprint and 5.5 times the speed.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
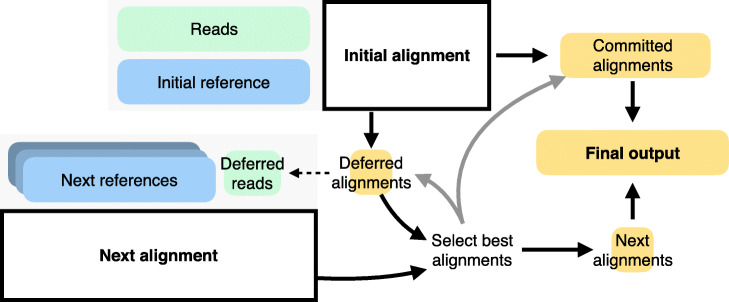
The reference flow workflow: Reads are aligned to reference genome in the first pass. Reads with high mapping quality alignments are “committed.” Unaligned reads or reads with low mapping quality are “deferred” and re-aligned to one or more additional references. The process can iterate, with similar logic for how reads are committed or deferred to another pass. Deferrals could follow the shape of an overall “reference flow graph.” Once all alignments are complete, alignments are merged. For a read aligning to more than one reference, only the best is reported, with ties broken arbitrarily. Alignments are translated (“lifted over”) to the coordinates of a standard reference like GRCh38
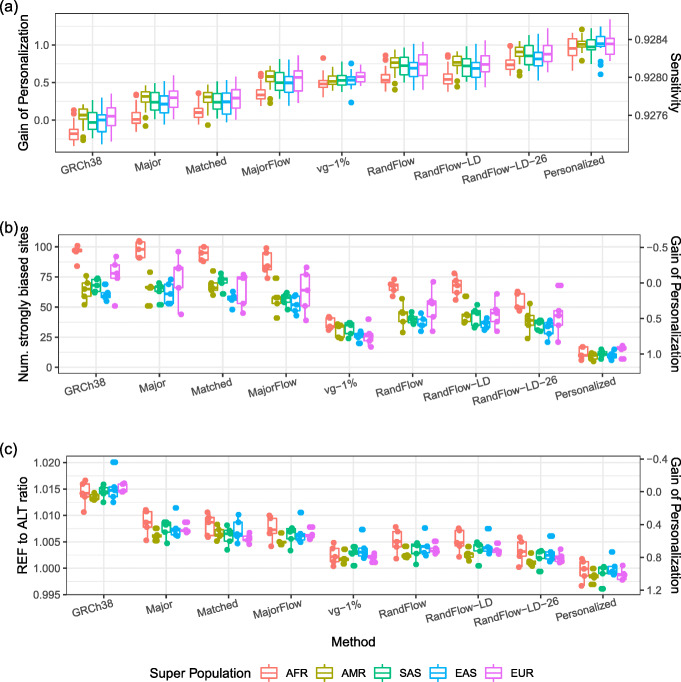
Alignment results using different methods. a Alignment sensitivity for 100 samples selected from the 1000 Genomes Project; 2 million reads are simulated from each sample. b The number of strongly biased heterozygous sites, and c the overall REF-to-ALT ratio for 25 samples; 20 million reads are simulated for each sample. The columns are sorted by median alignment sensitivity
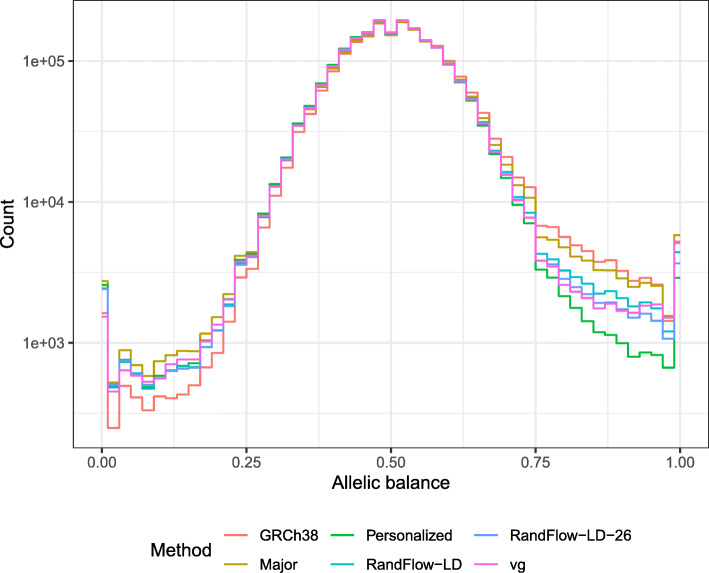
Histograms of allelic balance using a high-coverage real WGS dataset of individual NA12878 (SRR622457). Experiments are performed using GRCh38 (GRC), global major reference (Major), diploid personalized genome (Personalized), vg using alleles with frequency ≥ 10% (vg), reference flow using 1000-bp phased blocks with 5 super populations (RandFlow-LD) and reference flow using 1000-bp phased blocks with 26 populations (RandFlow-LD-26)
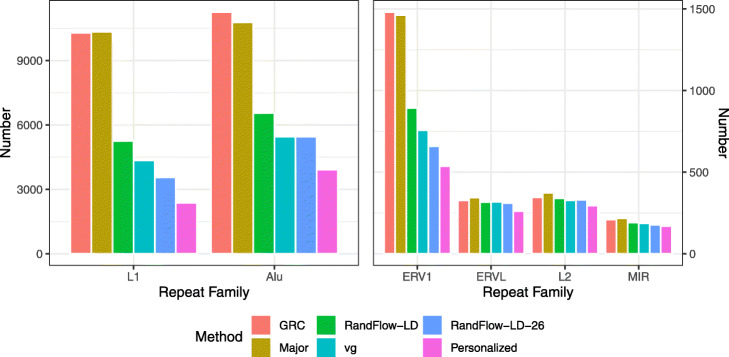
Number of strongly biased HET sites stratified by RepeatMasker class, after aligning single-end reads from SRR622457. HET sites are determined using 1000 Genomes Project calls for NA12878, the individual sequenced in SRR622457. RandFlow methods and vg reduce the number of biased sites substantially for L1, Alu, and ERV1. RandFlow-LD-26 reduces the number of biased sites most among the methods tested
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
