

Integrated genomic and transcriptomic analysis reveals unique characteristics of hepatic metastases and pro-metastatic role of complement C1q in pancreatic ductal adenocarcinoma
- PMID: 33397441
- PMCID: PMC7780398
- DOI: 10.1186/s13059-020-02222-w
Integrated genomic and transcriptomic analysis reveals unique characteristics of hepatic metastases and pro-metastatic role of complement C1q in pancreatic ductal adenocarcinoma
Abstract
Background: Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers due to its high metastasis rate in the liver. However, little is known about the molecular features of hepatic metastases due to difficulty in obtaining fresh tissues and low tumor cellularity.
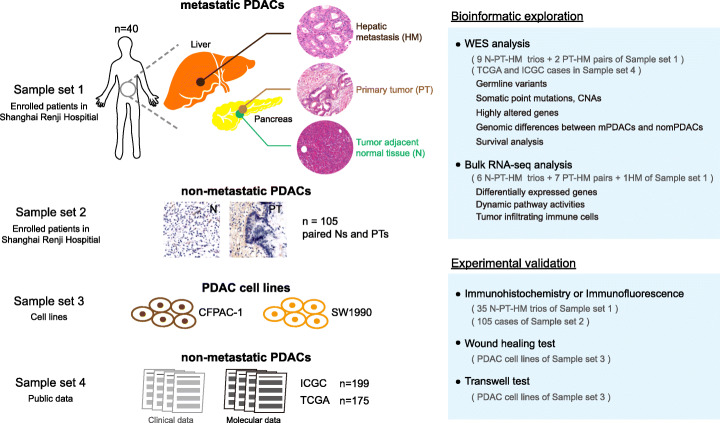
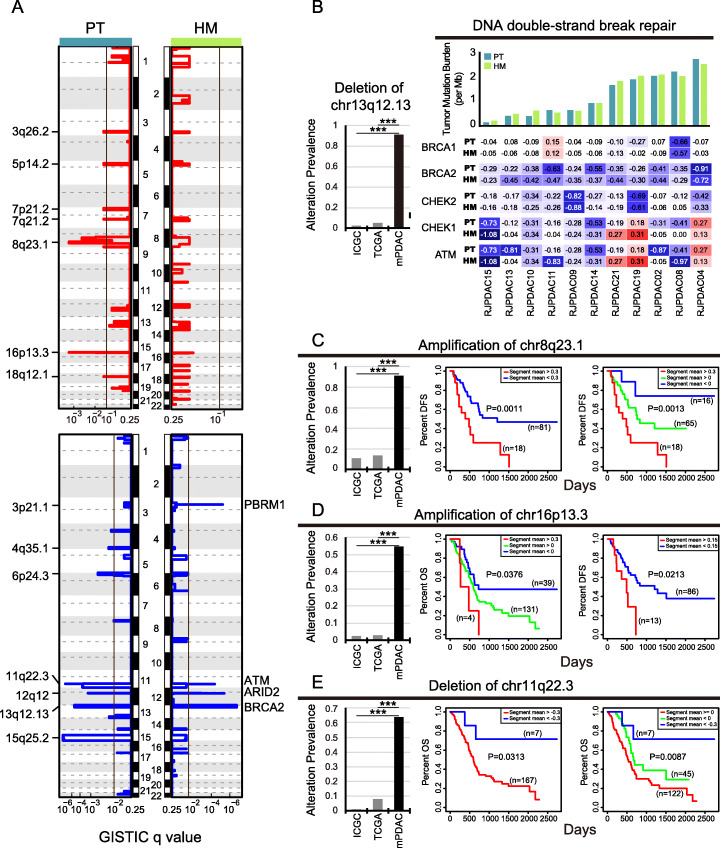
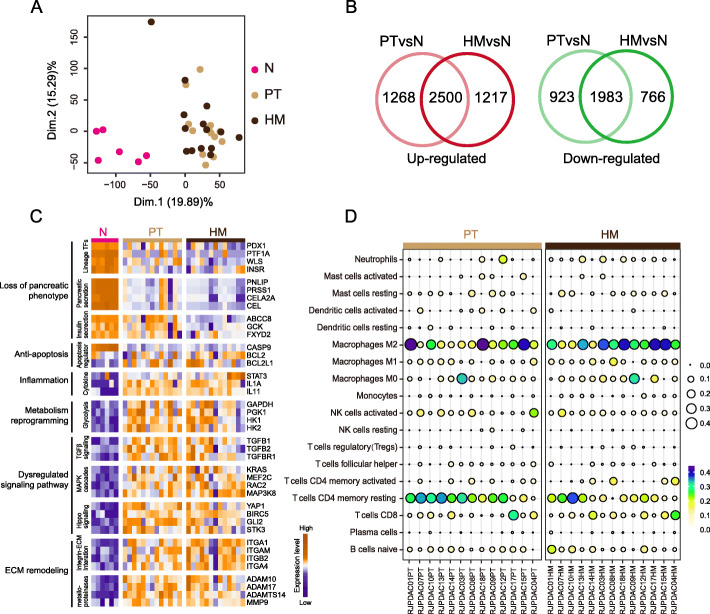
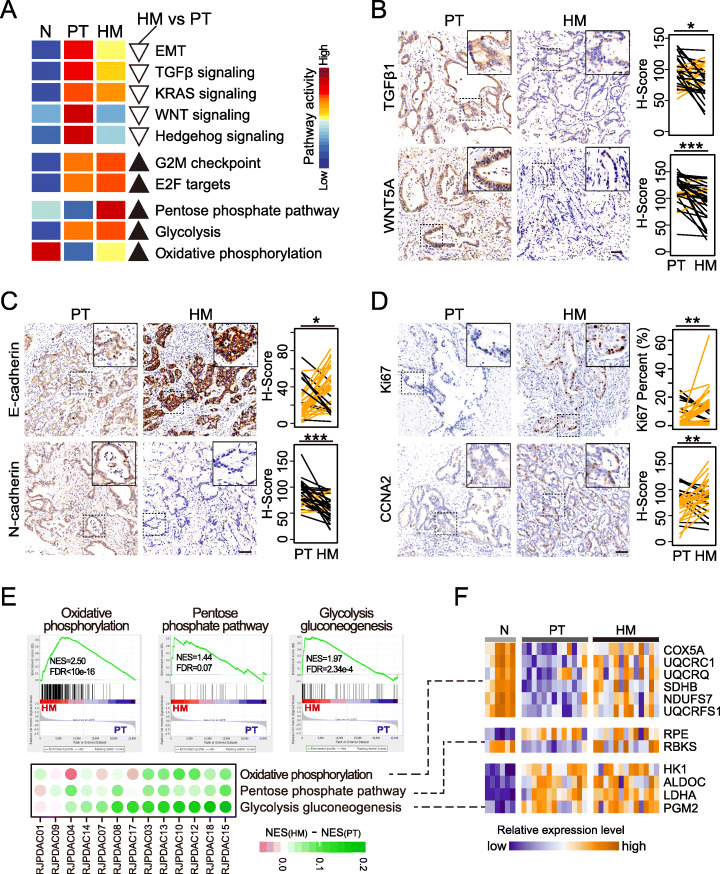
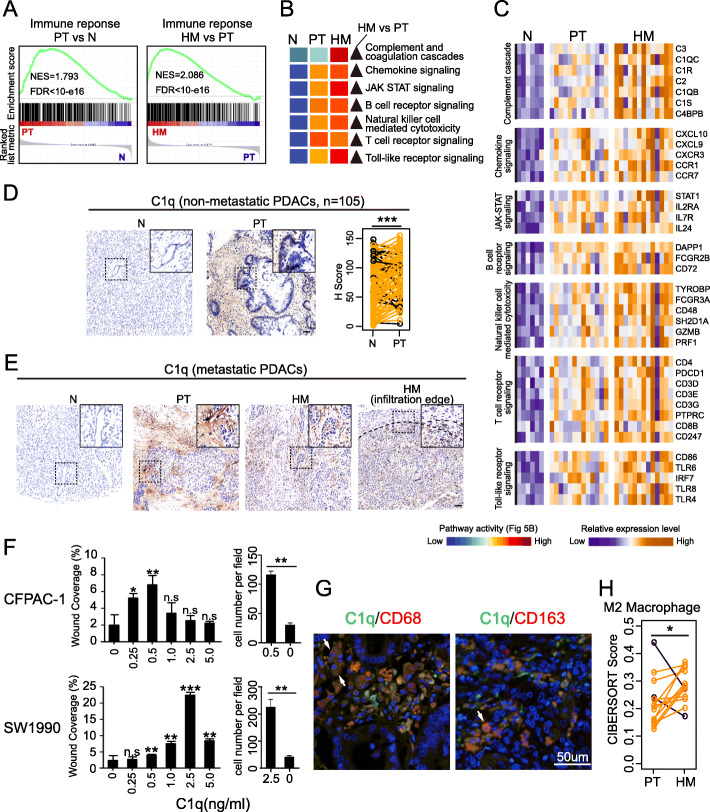
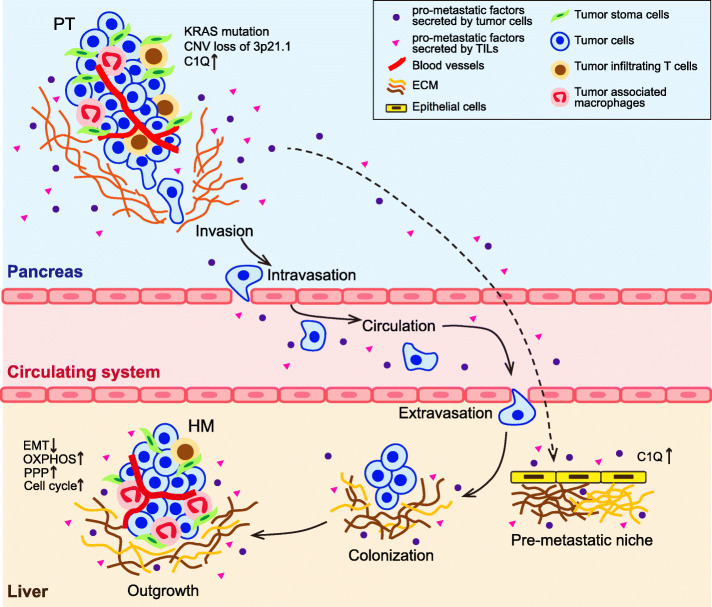
Results: We conduct exome sequencing and RNA sequencing for synchronous surgically resected primary tumors and the paired hepatic metastases from 17 hepatic oligometastatic pancreatic ductal adenocarcinoma and validate our findings in specimens from 35 of such cases. The comprehensive analysis of somatic mutations, copy number alterations, and gene expressions show high similarity between primary tumors and hepatic metastases. However, hepatic metastases also show unique characteristics, such as a higher degree of 3p21.1 loss, stronger abilities of proliferation, downregulation of epithelial to mesenchymal transition activity, and metabolic rewiring. More interesting, altered tumor microenvironments are observed in hepatic metastases, especially a higher proportion of tumor infiltrating M2 macrophage and upregulation of complement cascade. Further experiments demonstrate that expression of C1q increases in primary tumors and hepatic metastases, C1q is mainly produced by M2 macrophage, and C1q promotes migration and invasion of PDAC cells.
Conclusion: Taken together, we find potential factors that contribute to different stages of PDAC metastasis. Our study broadens the understanding of molecular mechanisms driving PDAC metastasis.
Keywords: C1q; Genomics; Hepatic metastasis; Pancreatic ductal adenocarcinoma; Transcriptomics; Tumor microenvironment.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med. 2009;133:413–422. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
