

scMC learns biological variation through the alignment of multiple single-cell genomics datasets
- PMID: 33397454
- PMCID: PMC7784288
- DOI: 10.1186/s13059-020-02238-2
scMC learns biological variation through the alignment of multiple single-cell genomics datasets
Abstract
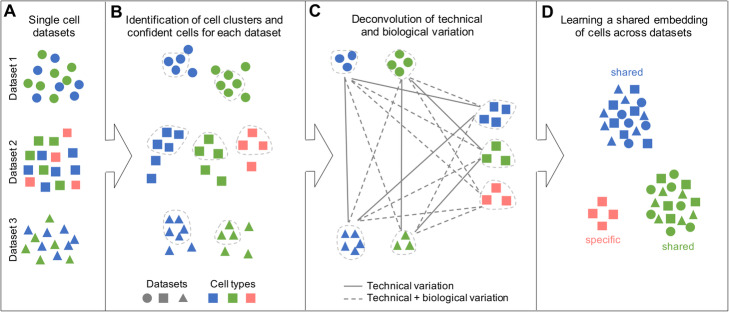
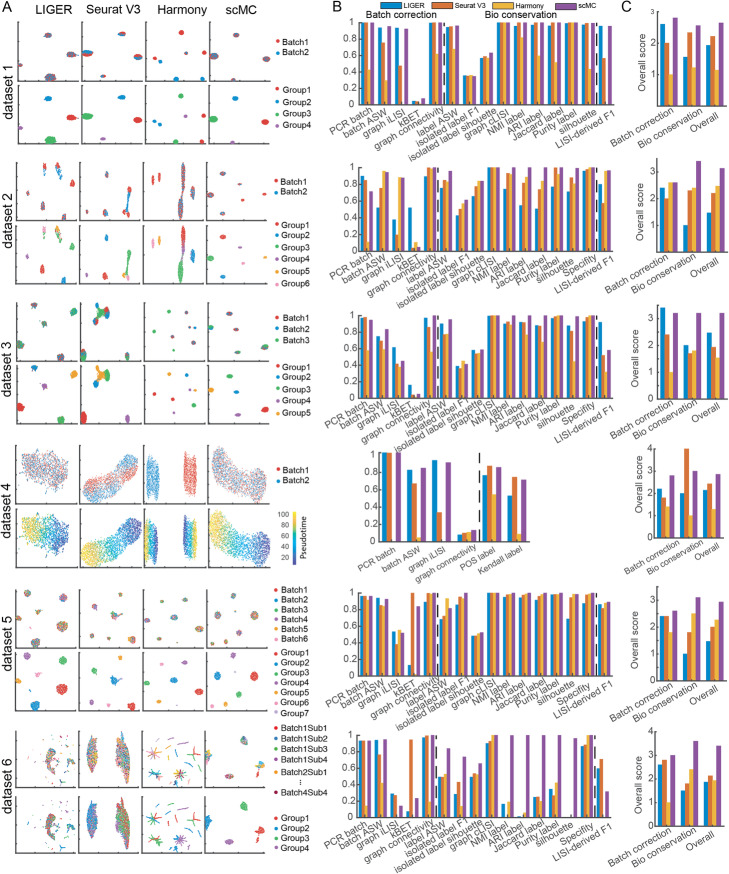
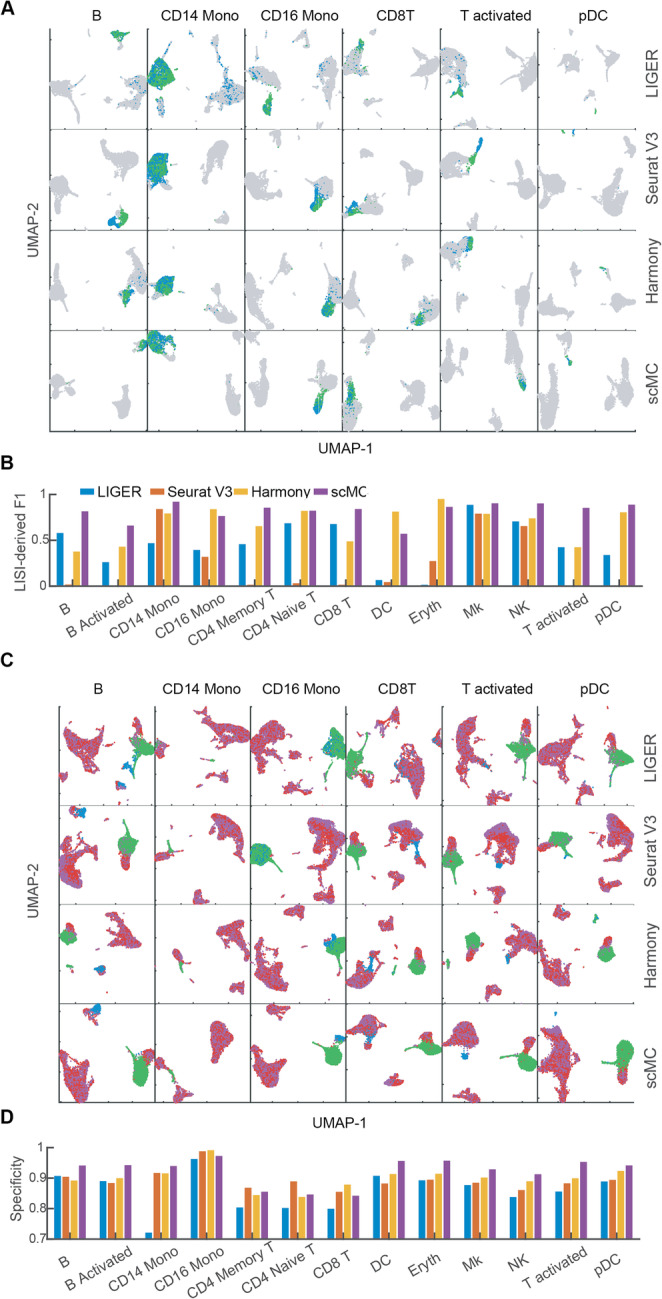
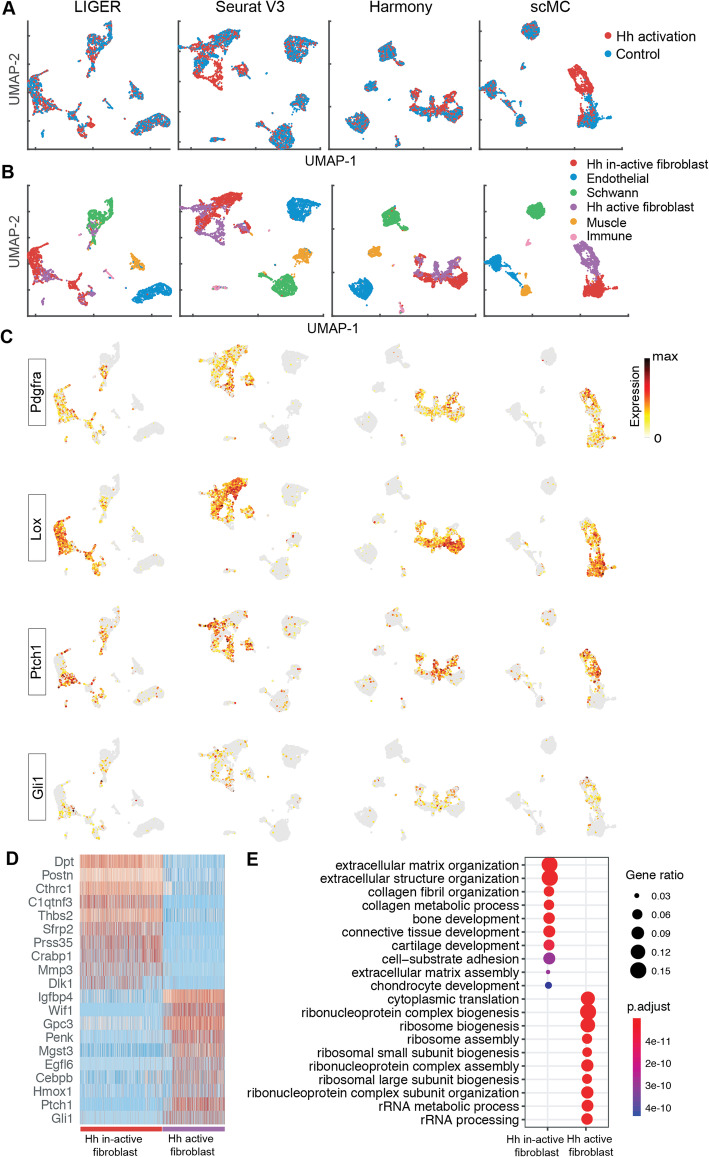
Distinguishing biological from technical variation is crucial when integrating and comparing single-cell genomics datasets across different experiments. Existing methods lack the capability in explicitly distinguishing these two variations, often leading to the removal of both variations. Here, we present an integration method scMC to remove the technical variation while preserving the intrinsic biological variation. scMC learns biological variation via variance analysis to subtract technical variation inferred in an unsupervised manner. Application of scMC to both simulated and real datasets from single-cell RNA-seq and ATAC-seq experiments demonstrates its capability of detecting context-shared and context-specific biological signals via accurate alignment.
Keywords: Single-cell genomics data, Data integration, Biological variation, Technical variation, Batch effect removal.
Conflict of interest statement
The authors declare no competing interests.
Figures
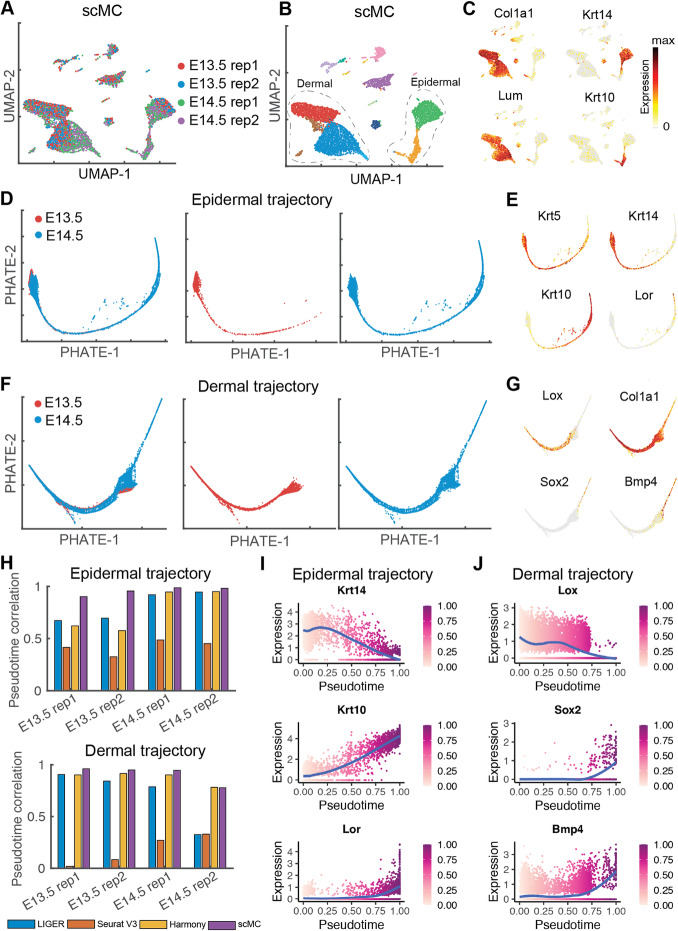
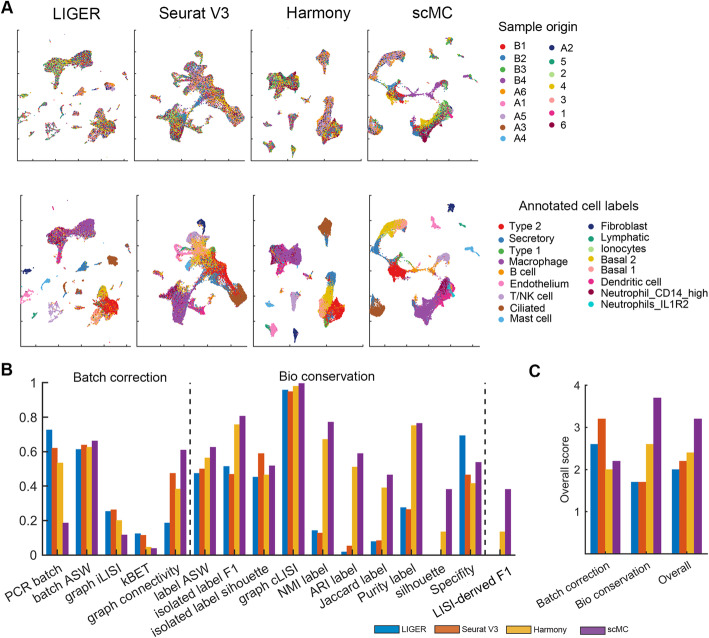
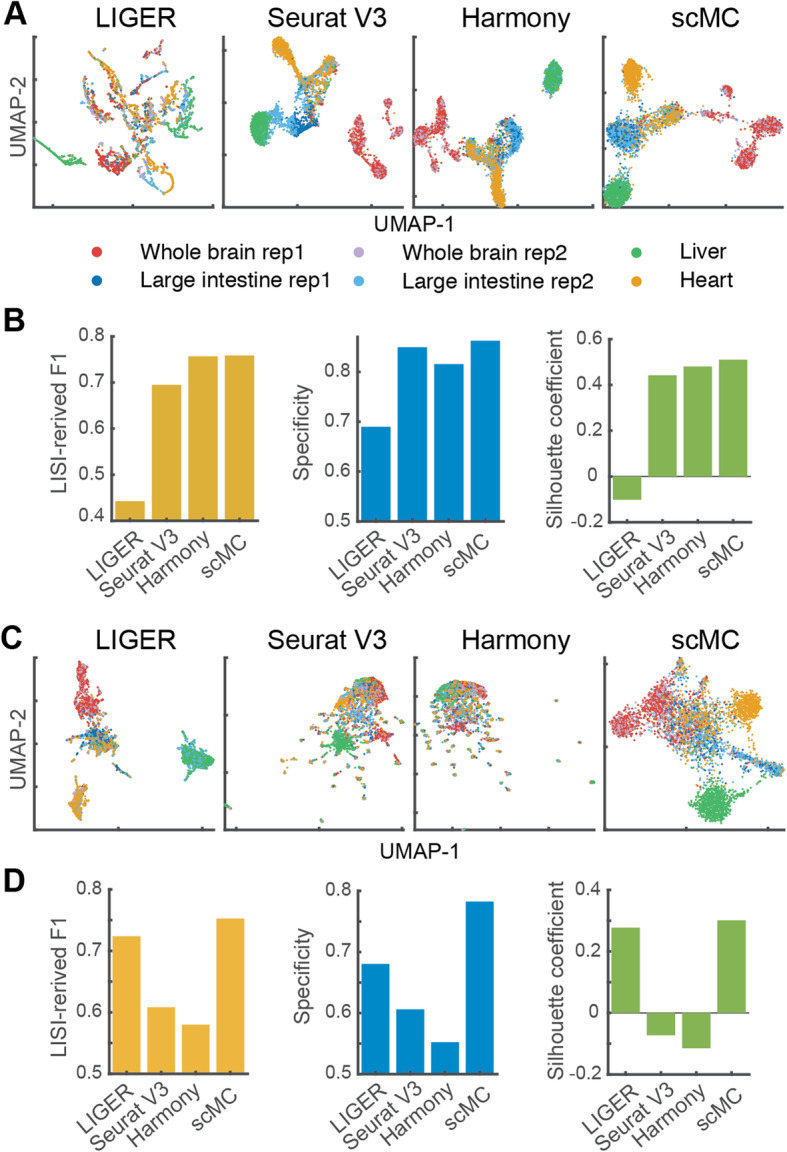
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
