

Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients
- PMID: 33397889
- PMCID: PMC7782482
- DOI: 10.1038/s41467-020-20162-8
Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients
Erratum in
-
Author Correction: Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients.Nat Commun. 2021 Feb 10;12(1):1048. doi: 10.1038/s41467-021-21285-2. Nat Commun. 2021. PMID: 33568659 Free PMC article. No abstract available.
Abstract
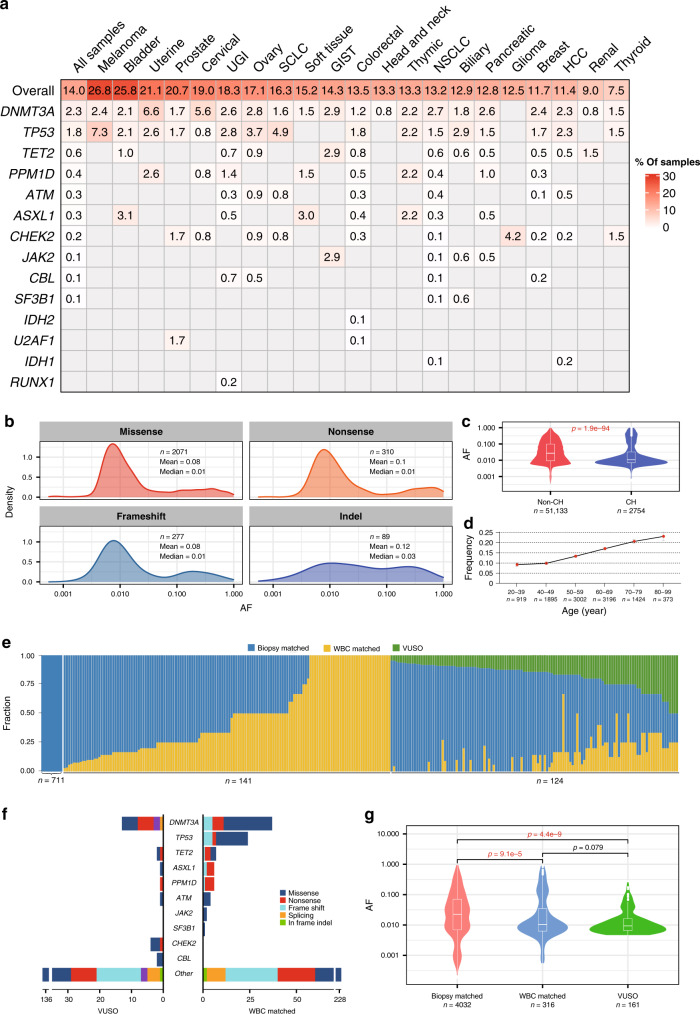
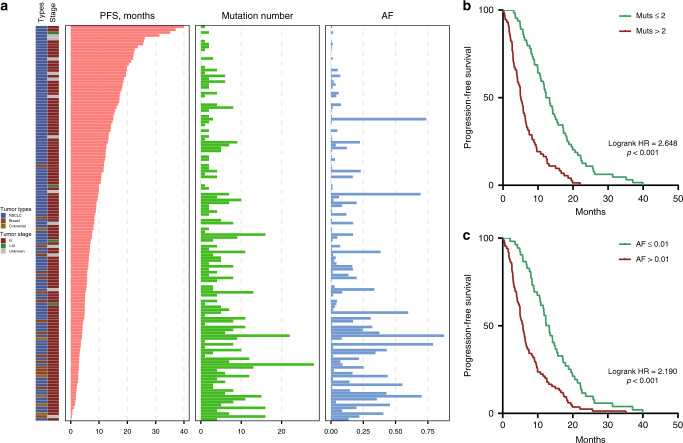
Circulating tumor DNA (ctDNA) provides a noninvasive approach to elucidate a patient's genomic landscape and actionable information. Here, we design a ctDNA-based study of over 10,000 pan-cancer Chinese patients. Using parallel sequencing between plasma and white blood cells, 14% of plasma cell-free DNA samples contain clonal hematopoiesis (CH) variants, for which detectability increases with age. After eliminating CH variants, ctDNA is detected in 73.5% of plasma samples, with small cell lung cancer (91.1%) and prostate cancer (87.9%) showing the highest detectability. The landscape of putative driver genes revealed by ctDNA profiling is similar to that in a tissue-based database (R2 = 0.87, p < 0.001) but also shows some discrepancies, such as higher EGFR (44.8% versus 25.2%) and lower KRAS (6.8% versus 27.2%) frequencies in non-small cell lung cancer, and a higher TP53 frequency in hepatocellular carcinoma (53.1% versus 28.6%). Up to 41.2% of plasma samples harbor drug-sensitive alterations. These findings may be helpful for identifying therapeutic targets and combined treatment strategies.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Allemani C, et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023–1075. doi: 10.1016/S0140-6736(17)33326-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
