

The role of water in host-guest interaction
- PMID: 33397926
- PMCID: PMC7782548
- DOI: 10.1038/s41467-020-20310-0
The role of water in host-guest interaction
Abstract
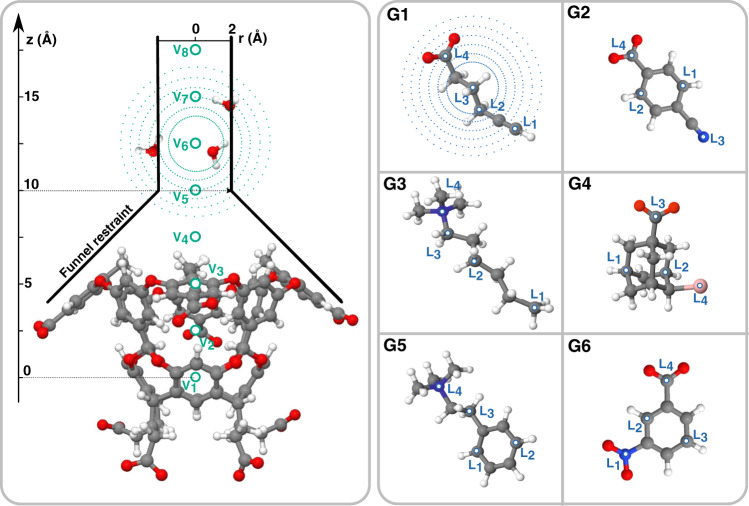
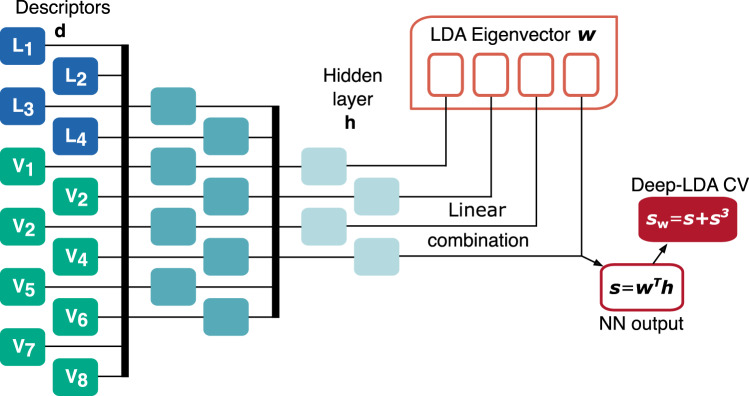
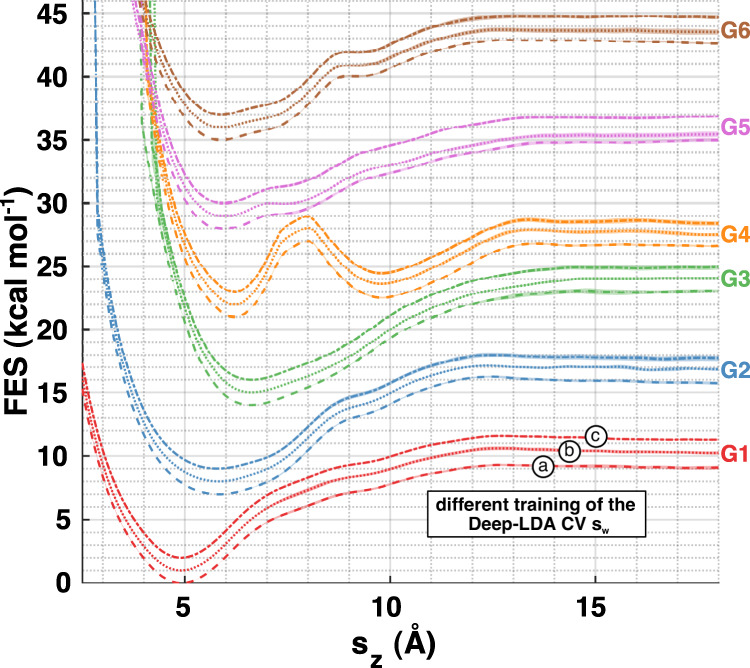
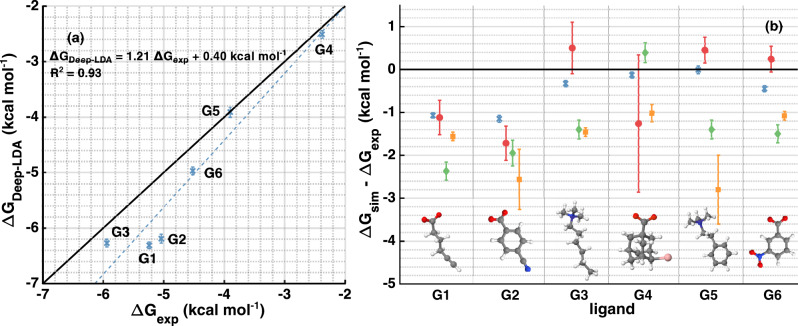
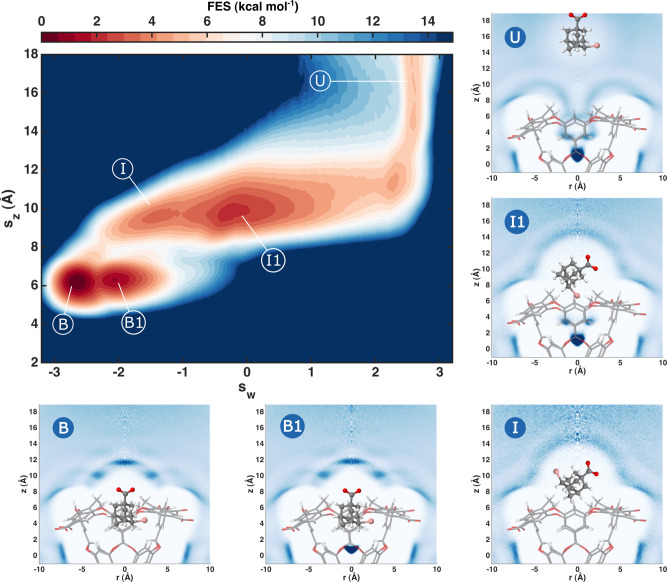
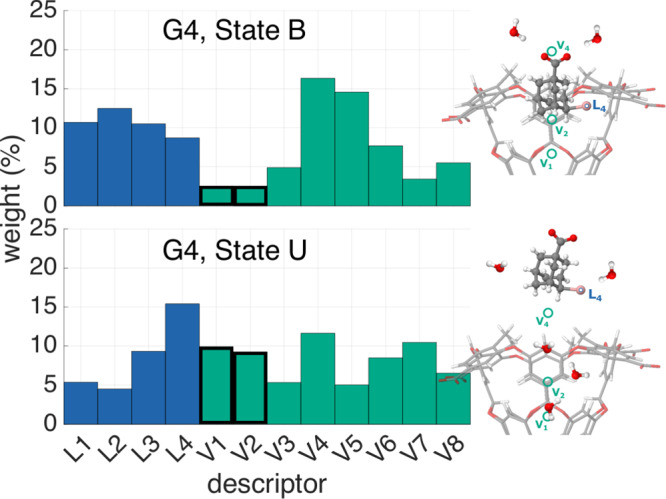
One of the main applications of atomistic computer simulations is the calculation of ligand binding free energies. The accuracy of these calculations depends on the force field quality and on the thoroughness of configuration sampling. Sampling is an obstacle in simulations due to the frequent appearance of kinetic bottlenecks in the free energy landscape. Very often this difficulty is circumvented by enhanced sampling techniques. Typically, these techniques depend on the introduction of appropriate collective variables that are meant to capture the system's degrees of freedom. In ligand binding, water has long been known to play a key role, but its complex behaviour has proven difficult to fully capture. In this paper we combine machine learning with physical intuition to build a non-local and highly efficient water-describing collective variable. We use it to study a set of host-guest systems from the SAMPL5 challenge. We obtain highly accurate binding free energies and good agreement with experiments. The role of water during the binding process is then analysed in some detail.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Bronowska, A. In Thermodynamics - Interaction Studies - Solids, Liquids and Gases Vol. i (InTech, 2011) https://www.intechopen.com/books/thermodynamics-interaction-studies-soli....
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
