

A practical guide to cancer subclonal reconstruction from DNA sequencing
- PMID: 33398189
- PMCID: PMC7867630
- DOI: 10.1038/s41592-020-01013-2
A practical guide to cancer subclonal reconstruction from DNA sequencing
Abstract
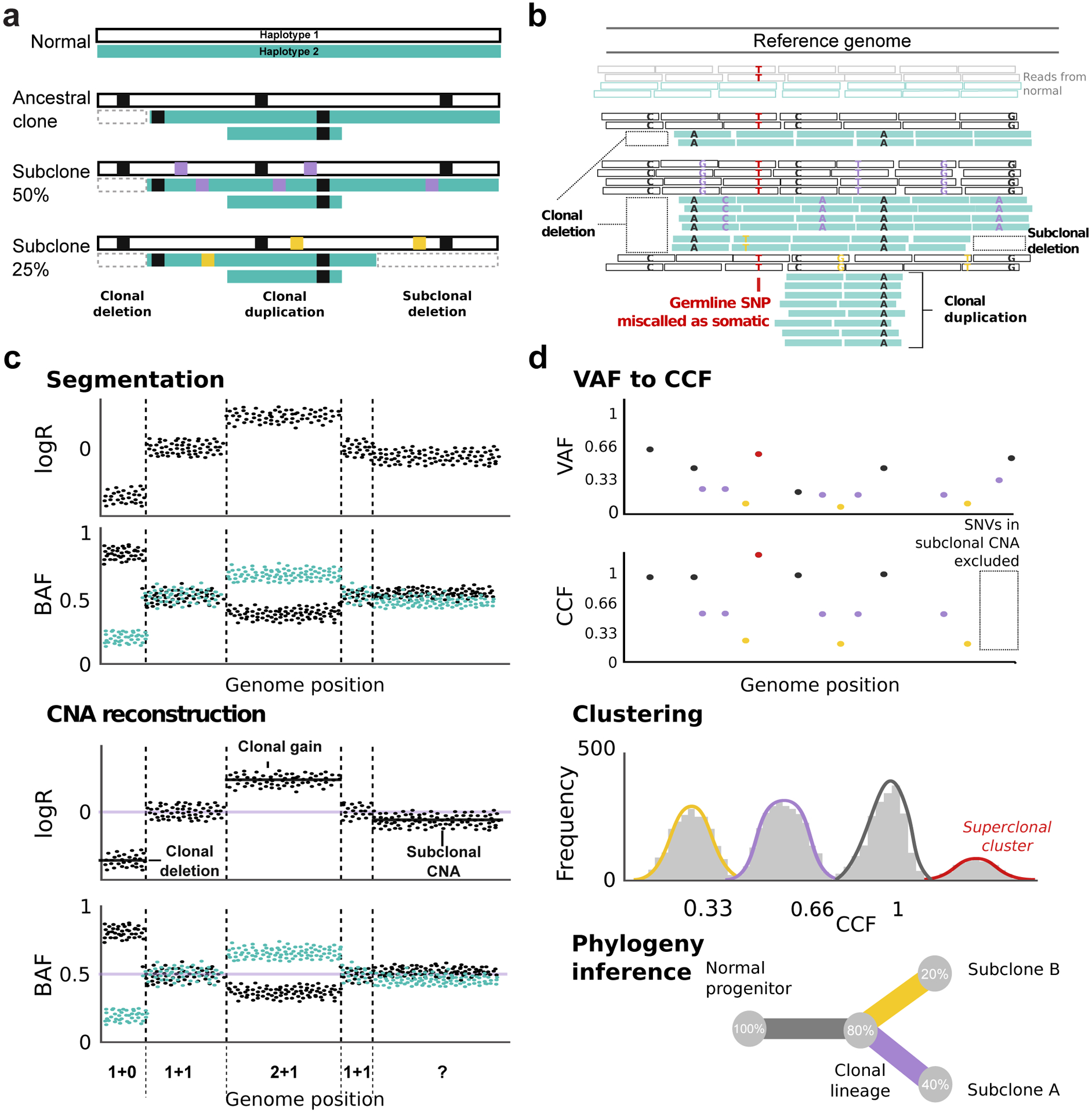
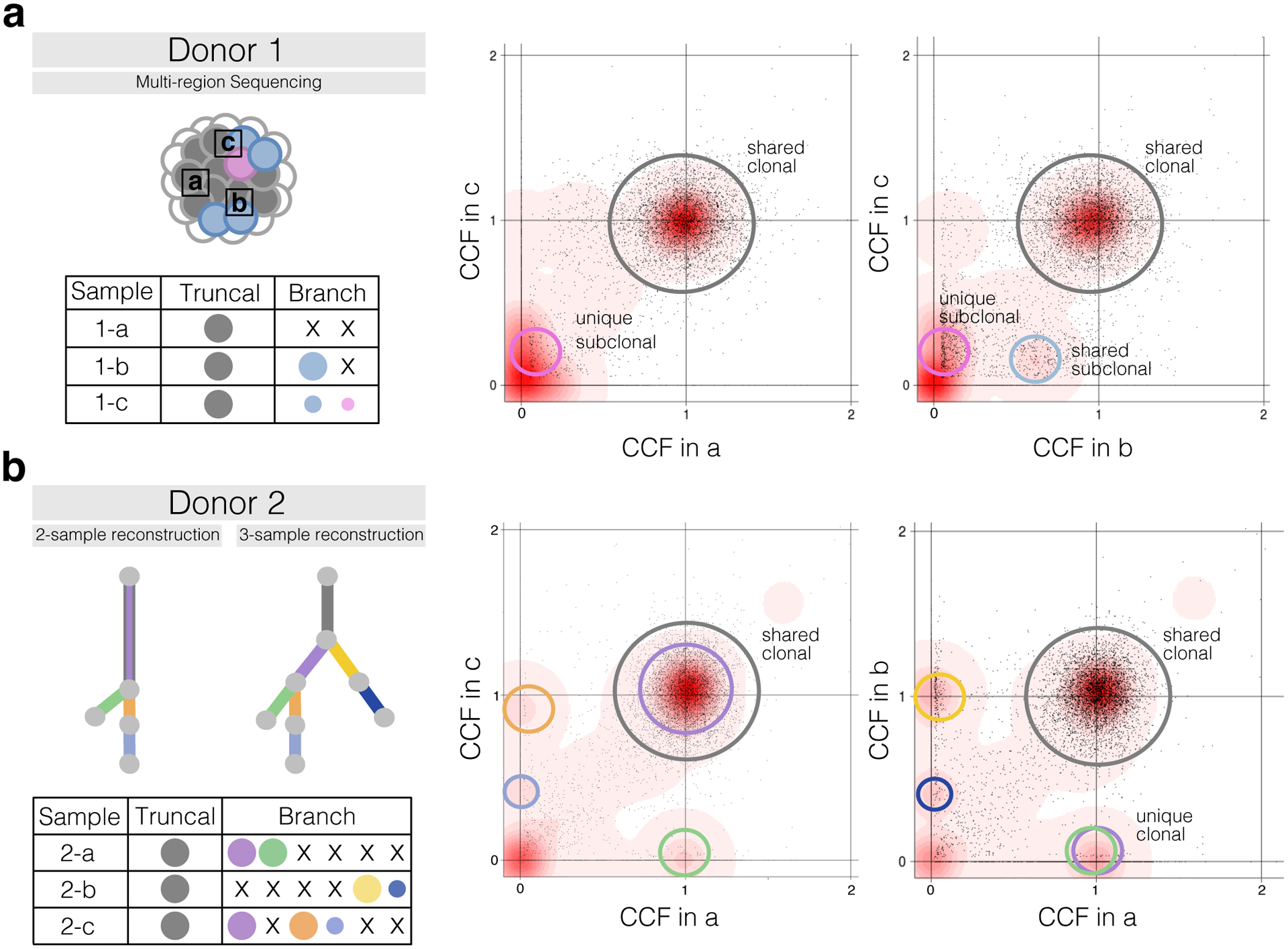
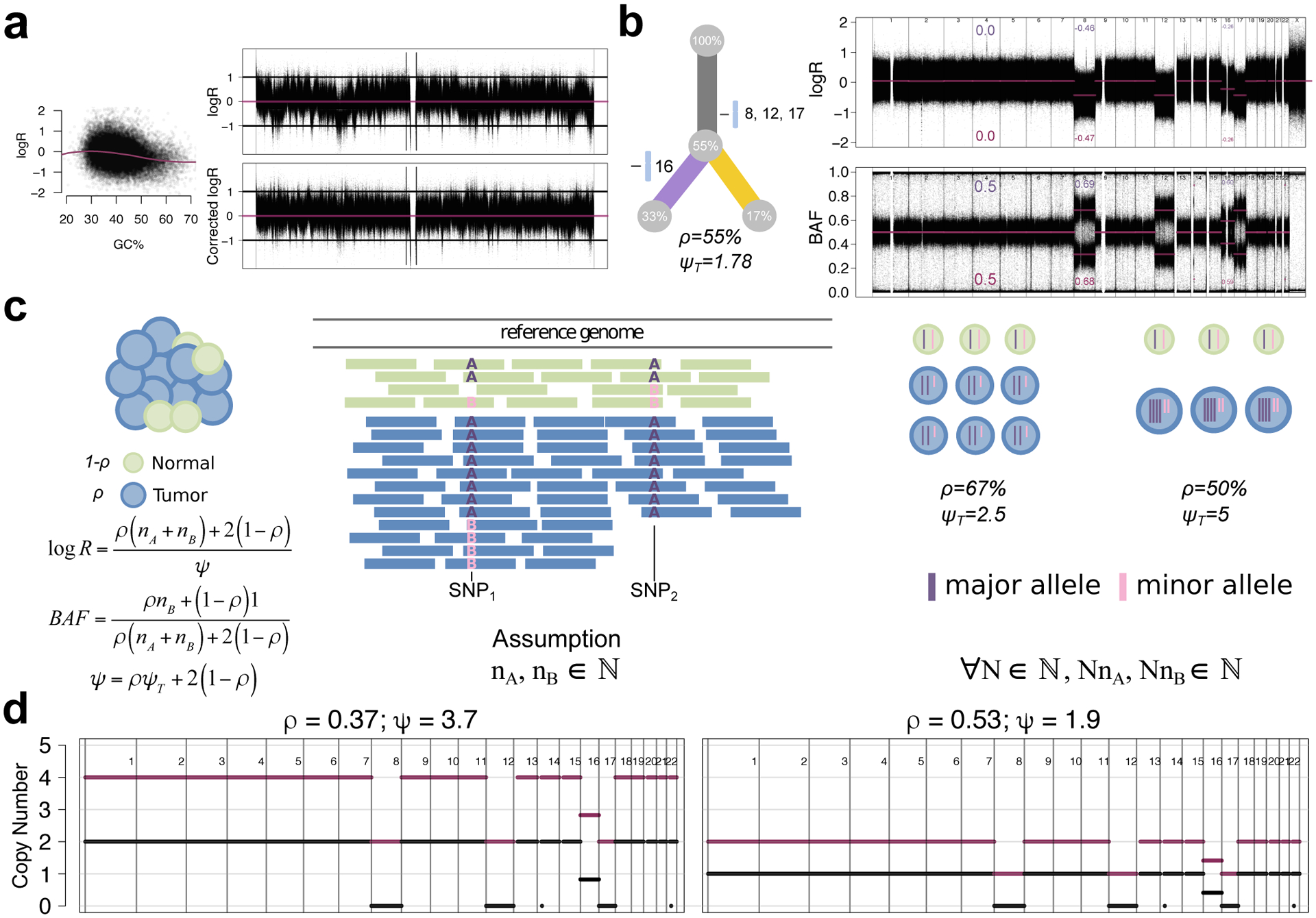
Subclonal reconstruction from bulk tumor DNA sequencing has become a pillar of cancer evolution studies, providing insight into the clonality and relative ordering of mutations and mutational processes. We provide an outline of the complex computational approaches used for subclonal reconstruction from single and multiple tumor samples. We identify the underlying assumptions and uncertainties in each step and suggest best practices for analysis and quality assessment. This guide provides a pragmatic resource for the growing user community of subclonal reconstruction methods.
Conflict of interest statement
Competing interests
P.C.B is a member of the Scientific Advisory Boards of BioSymetrics Inc. and Intersect Diagnostics Inc. M.T., A.S., A.G.D., M.N.L., J.W., D.C.W., Q.D.M., and P.V.L. declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
