

This is a preprint.
Neutralising antibodies in Spike mediated SARS-CoV-2 adaptation
- PMID: 33398302
- PMCID: PMC7781345
- DOI: 10.1101/2020.12.05.20241927
Neutralising antibodies in Spike mediated SARS-CoV-2 adaptation
Update in
-
SARS-CoV-2 evolution during treatment of chronic infection.Nature. 2021 Apr;592(7853):277-282. doi: 10.1038/s41586-021-03291-y. Epub 2021 Feb 5. Nature. 2021. PMID: 33545711 Free PMC article.
Abstract
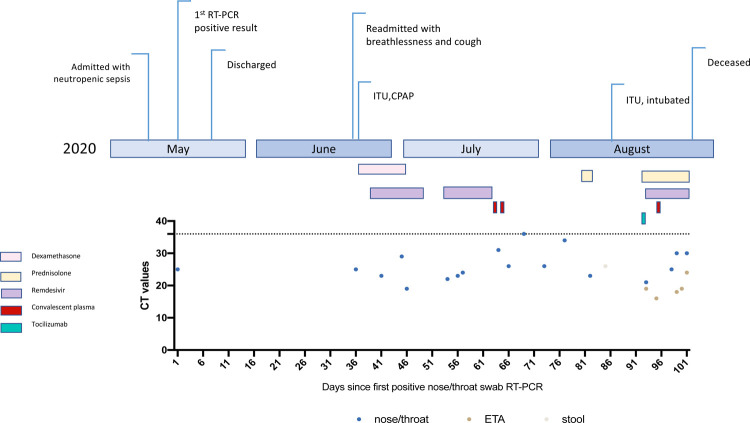
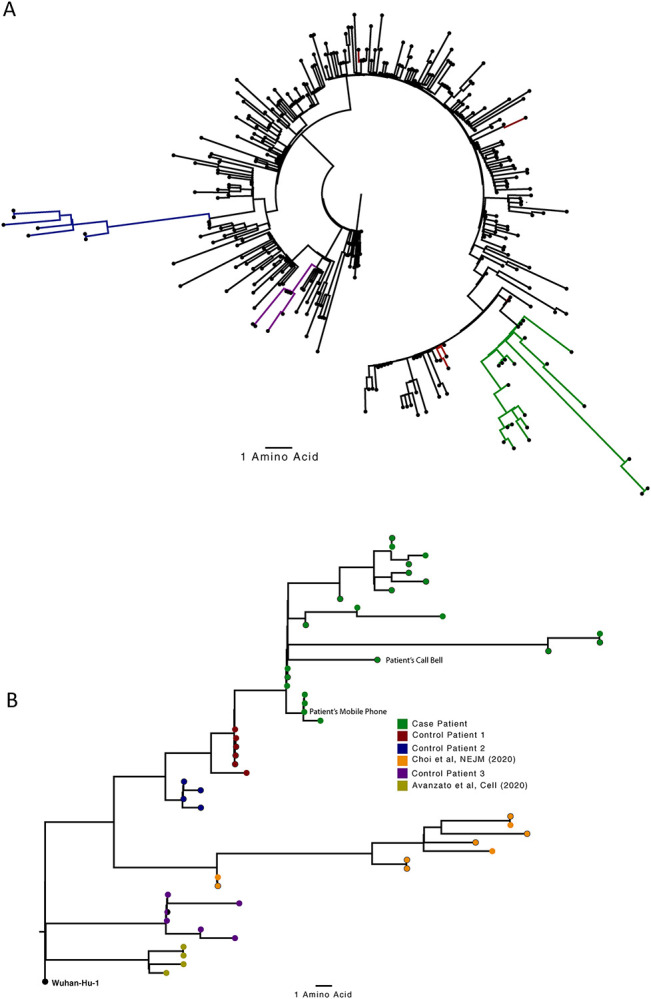
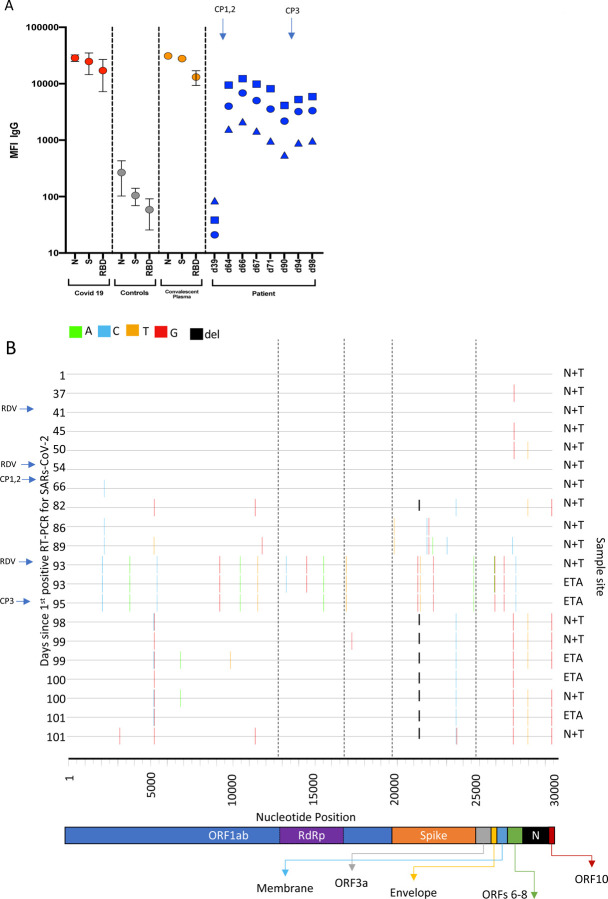
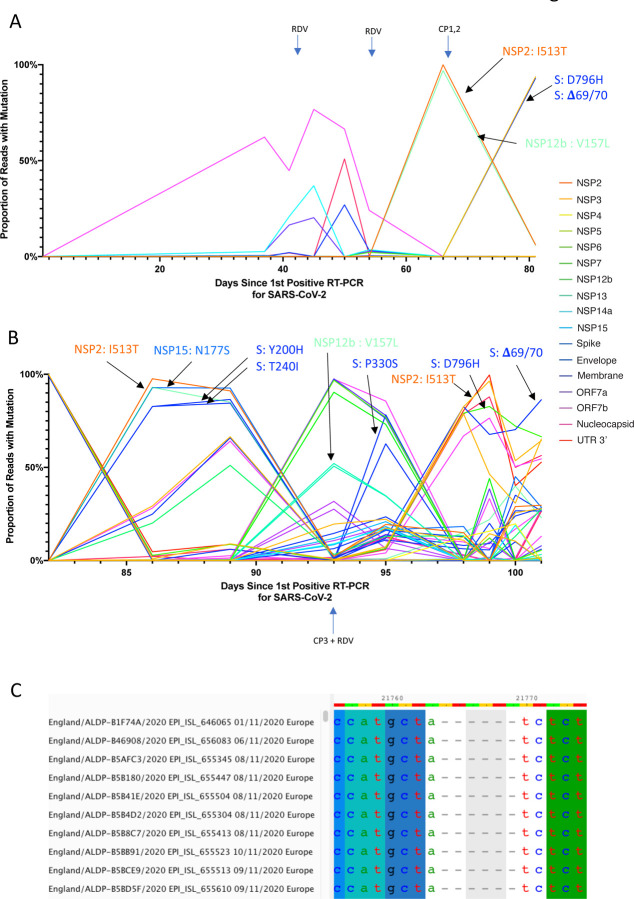
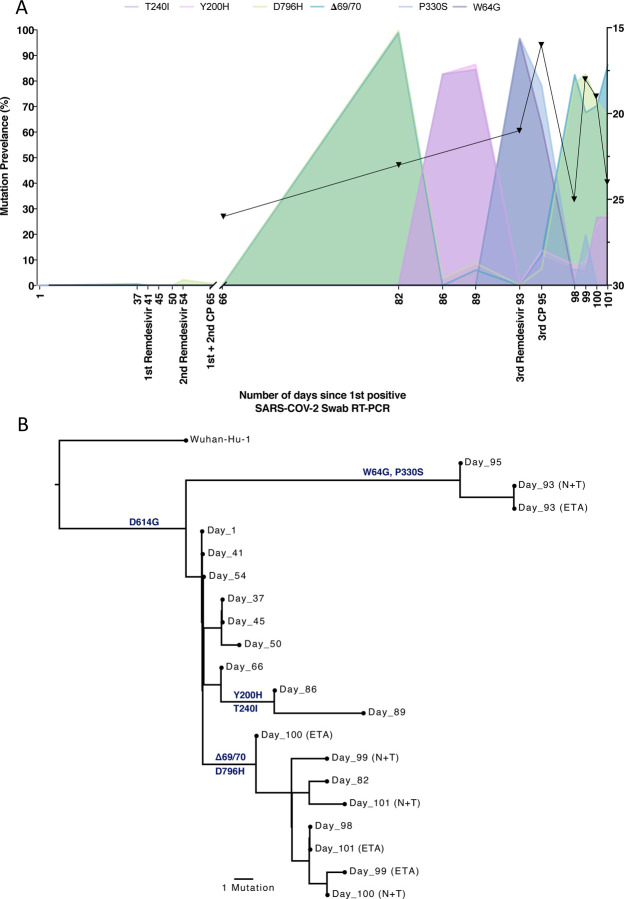
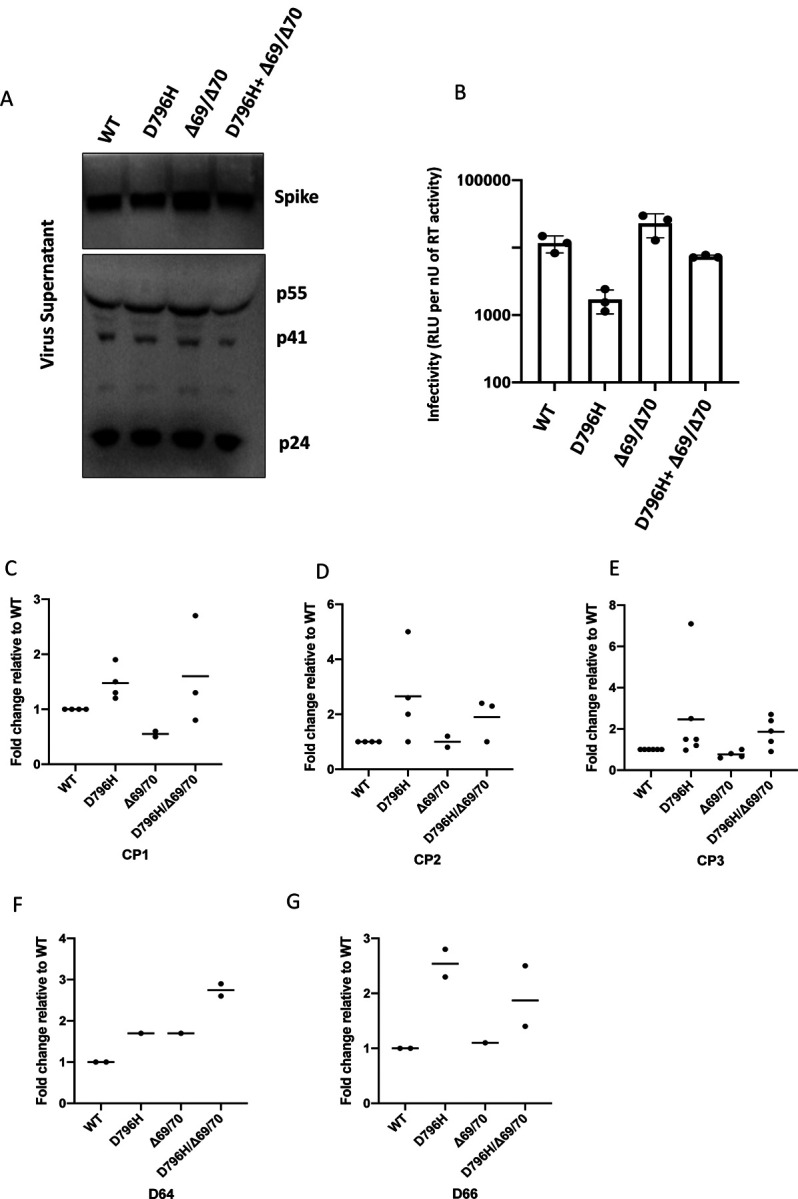
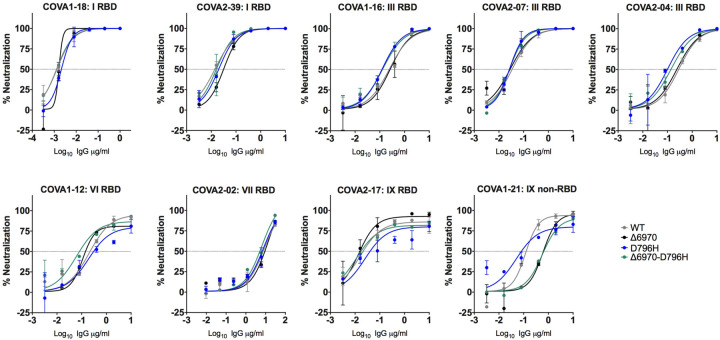
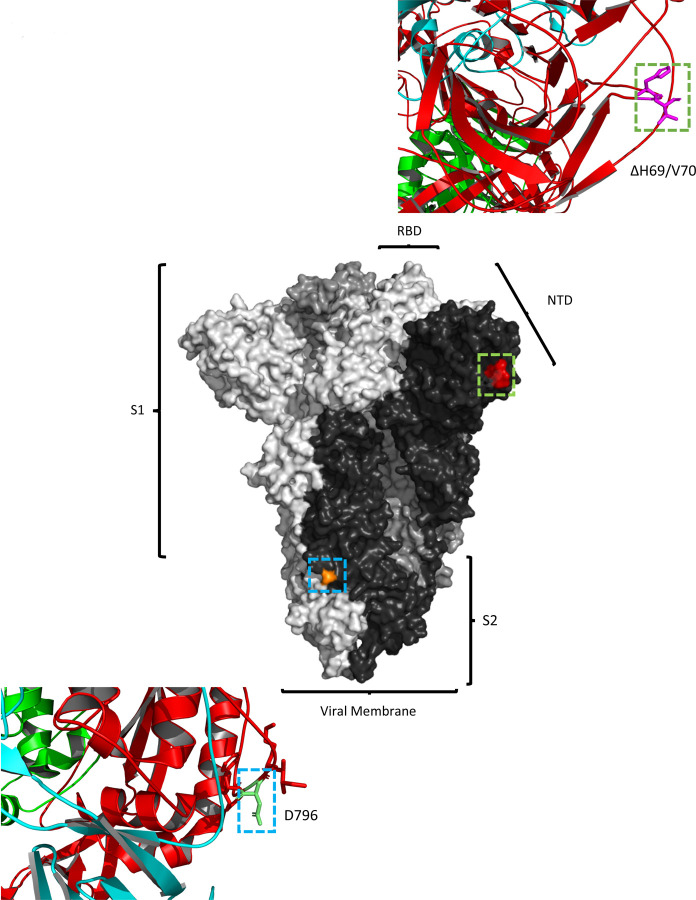
SARS-CoV-2 Spike protein is critical for virus infection via engagement of ACE2, and amino acid variation in Spike is increasingly appreciated. Given both vaccines and therapeutics are designed around Wuhan-1 Spike, this raises the theoretical possibility of virus escape, particularly in immunocompromised individuals where prolonged viral replication occurs. Here we report chronic SARS-CoV-2 with reduced sensitivity to neutralising antibodies in an immune suppressed individual treated with convalescent plasma, generating whole genome ultradeep sequences by both short and long read technologies over 23 time points spanning 101 days. Although little change was observed in the overall viral population structure following two courses of remdesivir over the first 57 days, N501Y in Spike was transiently detected at day 55 and V157L in RdRp emerged. However, following convalescent plasma we observed large, dynamic virus population shifts, with the emergence of a dominant viral strain bearing D796H in S2 and ΔH69/ΔV70 in the S1 N-terminal domain NTD of the Spike protein. As passively transferred serum antibodies diminished, viruses with the escape genotype diminished in frequency, before returning during a final, unsuccessful course of convalescent plasma. In vitro, the Spike escape double mutant bearing ΔH69/ΔV70 and D796H conferred decreased sensitivity to convalescent plasma, whilst maintaining infectivity similar to wild type. D796H appeared to be the main contributor to decreased susceptibility, but incurred an infectivity defect. The ΔH69/ΔV70 single mutant had two-fold higher infectivity compared to wild type and appeared to compensate for the reduced infectivity of D796H. Consistent with the observed mutations being outside the RBD, monoclonal antibodies targeting the RBD were not impacted by either or both mutations, but a non RBD binding monoclonal antibody was less potent against ΔH69/ΔV70 and the double mutant. These data reveal strong selection on SARS-CoV-2 during convalescent plasma therapy associated with emergence of viral variants with reduced susceptibility to neutralising antibodies.
Keywords: COVID-19; Convalescent plasma; SARS-CoV-2; antibody escape; evasion; immune suppression; mutation; neutralising antibodies; resistance.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous