

Targeting the APP-Mint2 Protein-Protein Interaction with a Peptide-Based Inhibitor Reduces Amyloid-β Formation
- PMID: 33398998
- PMCID: PMC8176898
- DOI: 10.1021/jacs.0c10696
Targeting the APP-Mint2 Protein-Protein Interaction with a Peptide-Based Inhibitor Reduces Amyloid-β Formation
Abstract
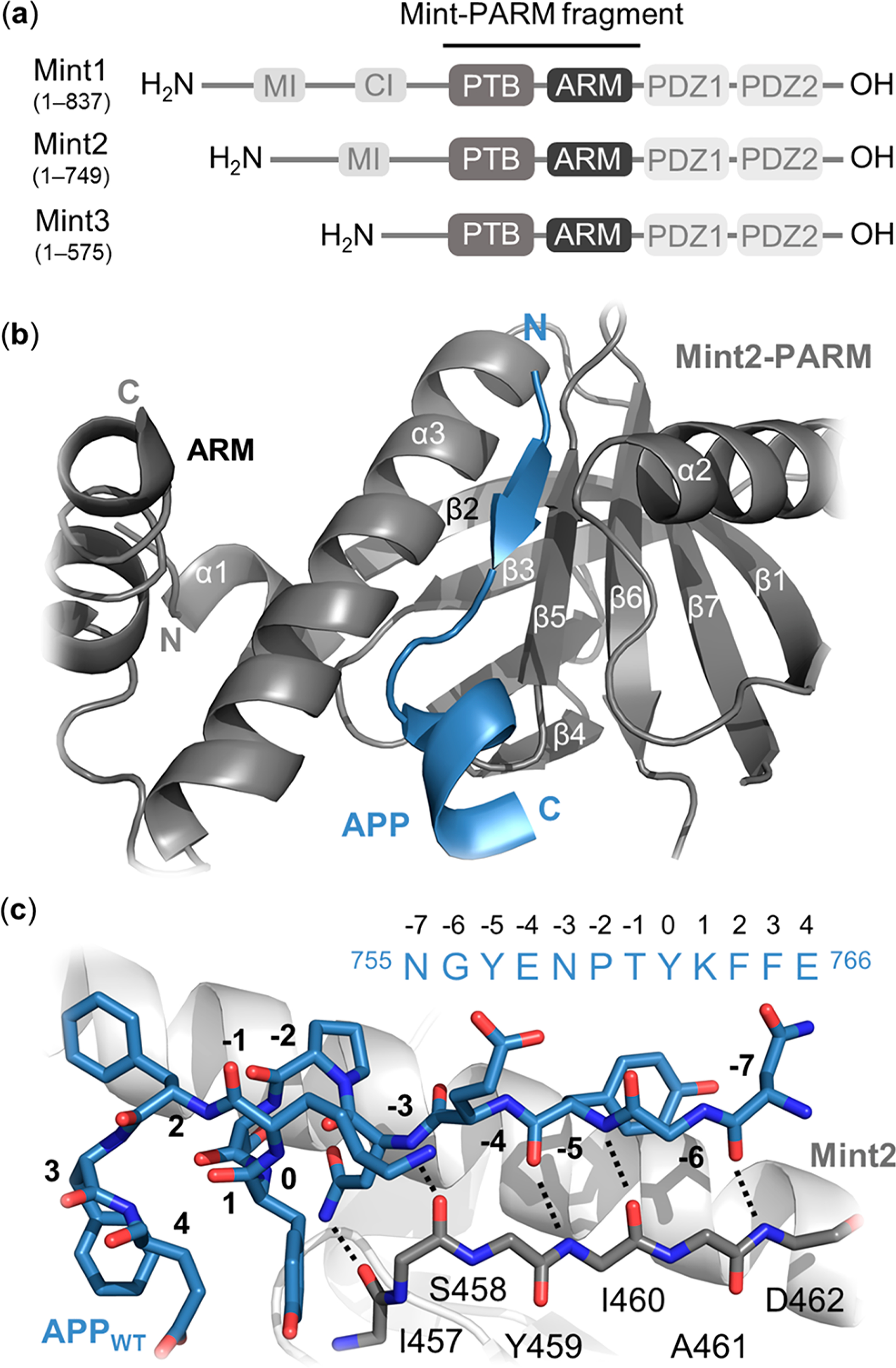
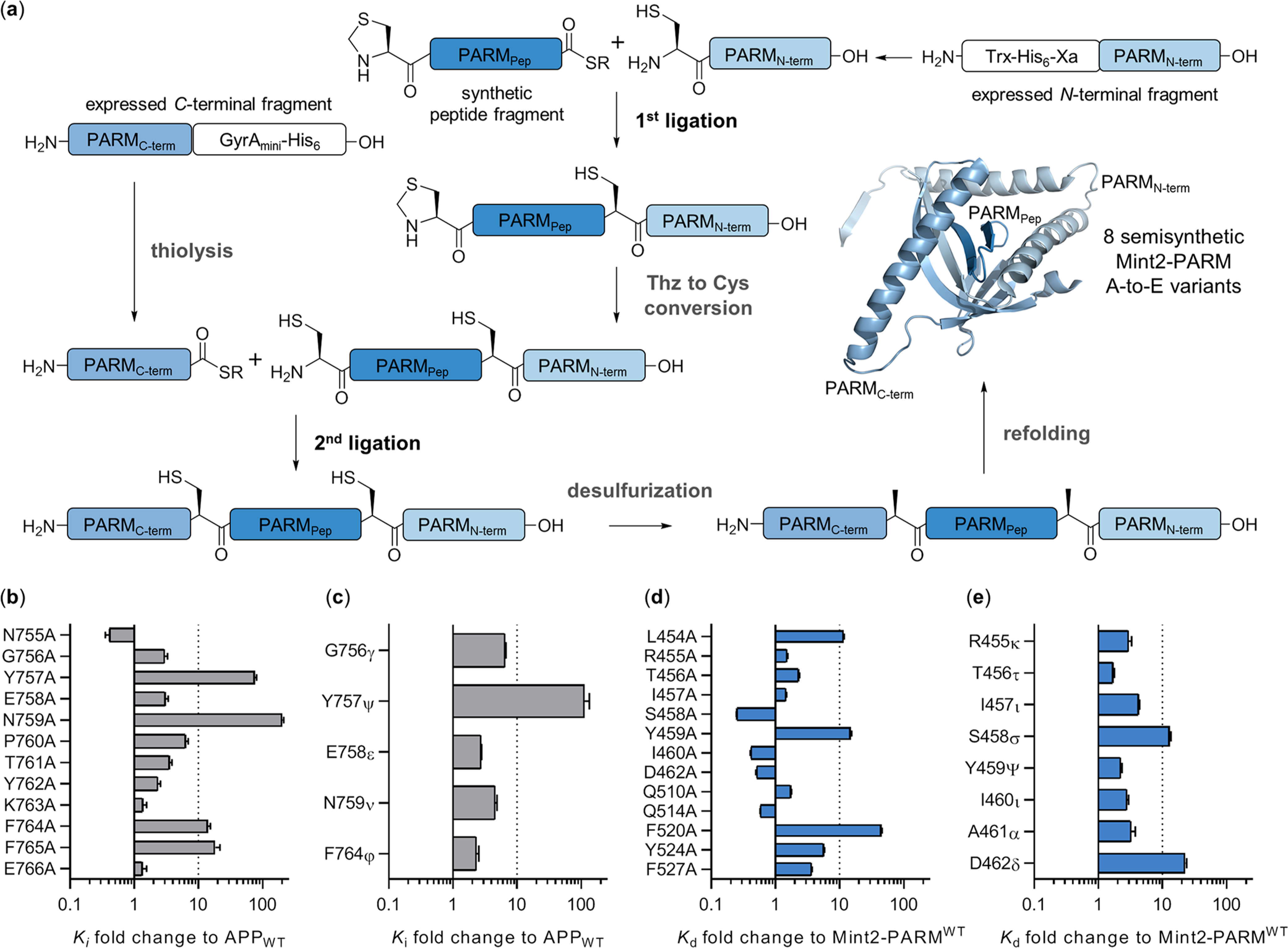
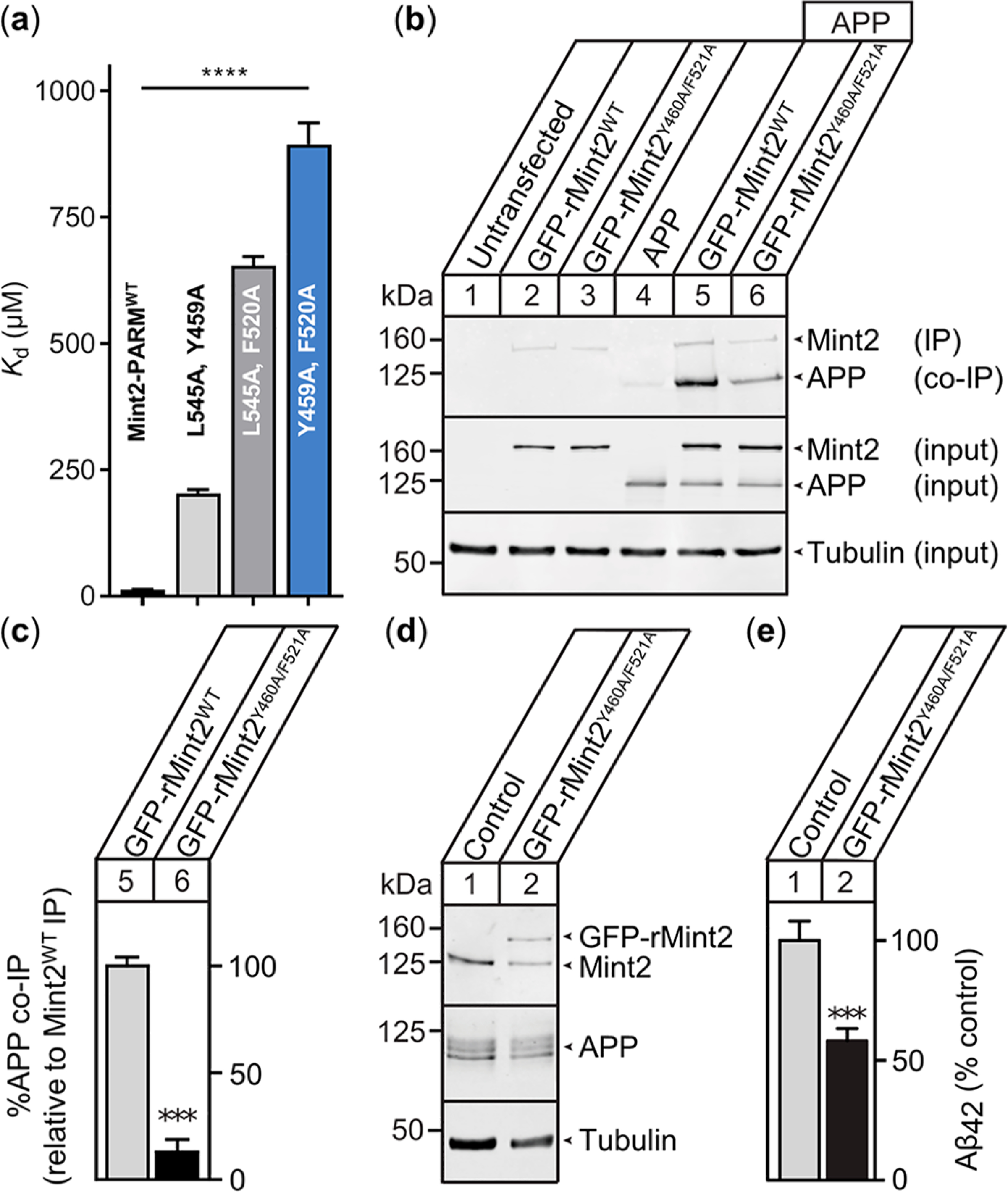
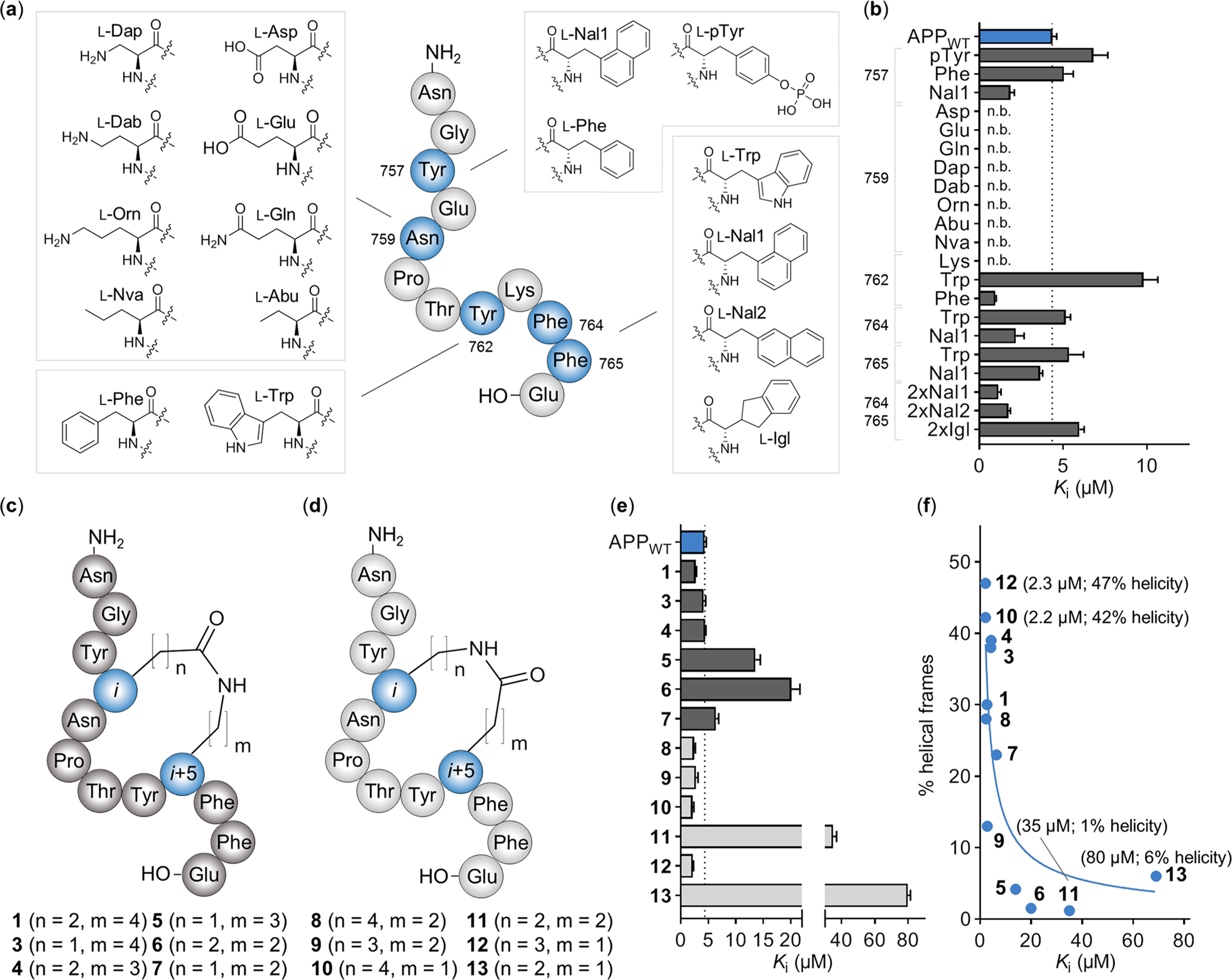
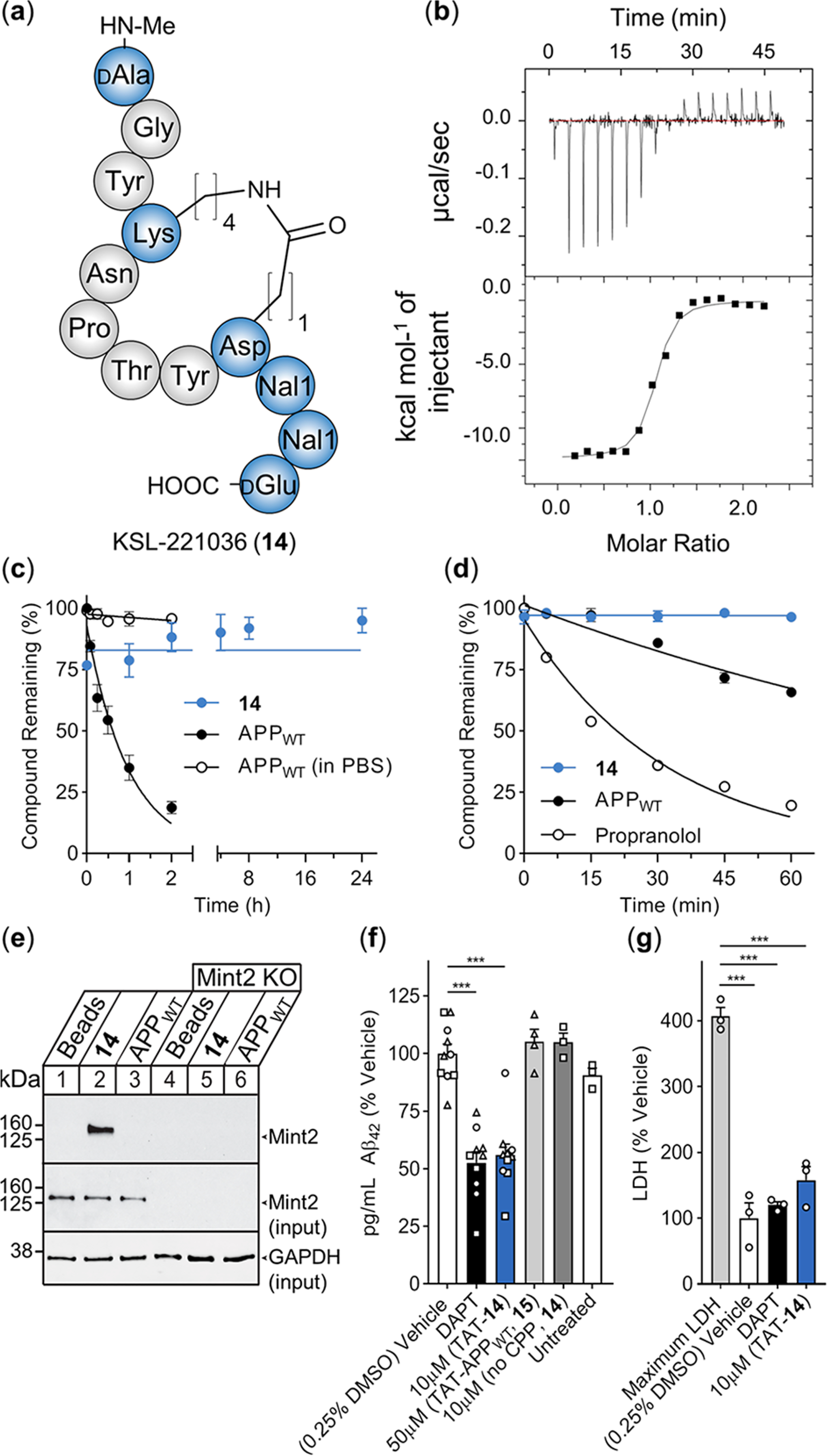
There is an urgent need for novel therapeutic approaches to treat Alzheimer's disease (AD) with the ability to both alleviate the clinical symptoms and halt the progression of the disease. AD is characterized by the accumulation of amyloid-β (Aβ) peptides which are generated through the sequential proteolytic cleavage of the amyloid precursor protein (APP). Previous studies reported that Mint2, a neuronal adaptor protein binding both APP and the γ-secretase complex, affects APP processing and formation of pathogenic Aβ. However, there have been contradicting results concerning whether Mint2 has a facilitative or suppressive effect on Aβ generation. Herein, we deciphered the APP-Mint2 protein-protein interaction (PPI) via extensive probing of both backbone H-bond and side-chain interactions. We also developed a proteolytically stable, high-affinity peptide targeting the APP-Mint2 interaction. We found that both an APP binding-deficient Mint2 variant and a cell-permeable PPI inhibitor significantly reduced Aβ42 levels in a neuronal in vitro model of AD. Together, these findings demonstrate a facilitative role of Mint2 in Aβ formation, and the combination of genetic and pharmacological approaches suggests that targeting Mint2 is a promising therapeutic strategy to reduce pathogenic Aβ levels.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Wang J; Gu BJ; Masters CL; Wang YJ A systemic view of Alzheimer disease - insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13 (10), 612–623. - PubMed
-
- Müller UC; Deller T; Korte M Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18 (5), 281–298. - PubMed
-
- Haass C; Selkoe DJ Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8 (2), 101–112. - PubMed
-
- Panza F; Lozupone M; Logroscino G; Imbimbo BP A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15 (2), 73–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
