

Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery
- PMID: 33400914
- PMCID: PMC7904613
- DOI: 10.1016/j.devcel.2020.12.015
Single-cell RNA sequencing of developing maize ears facilitates functional analysis and trait candidate gene discovery
Abstract
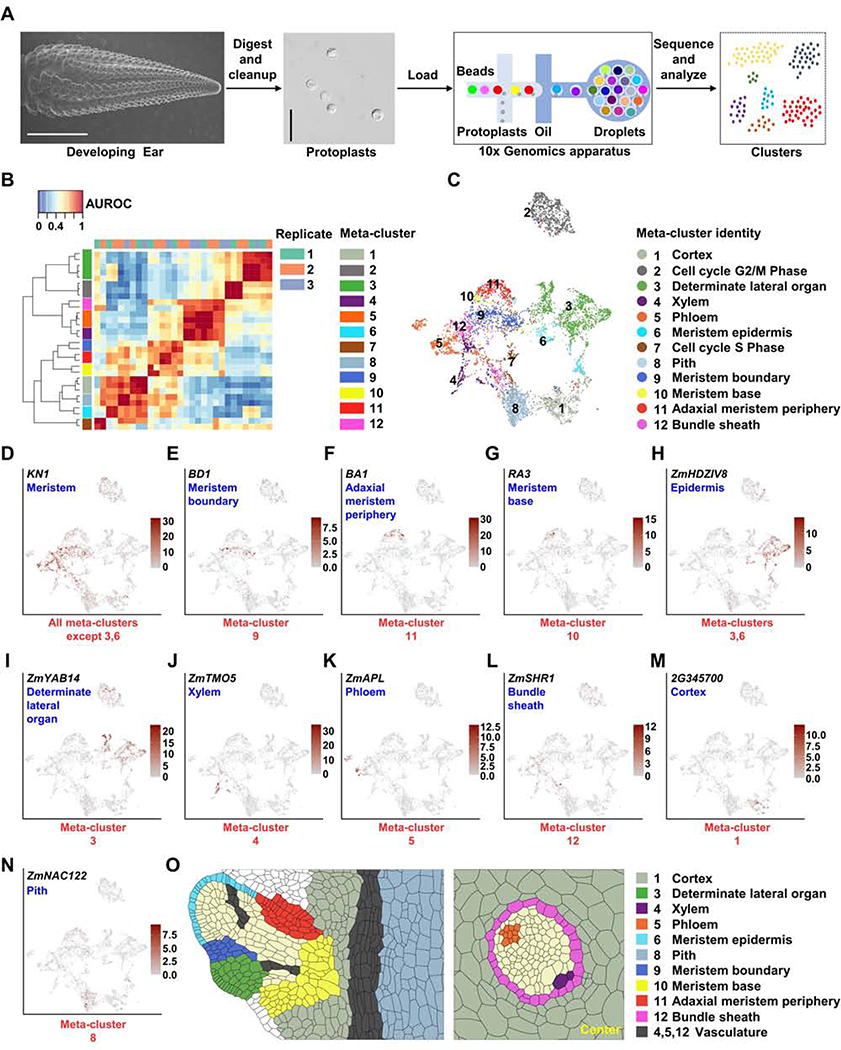
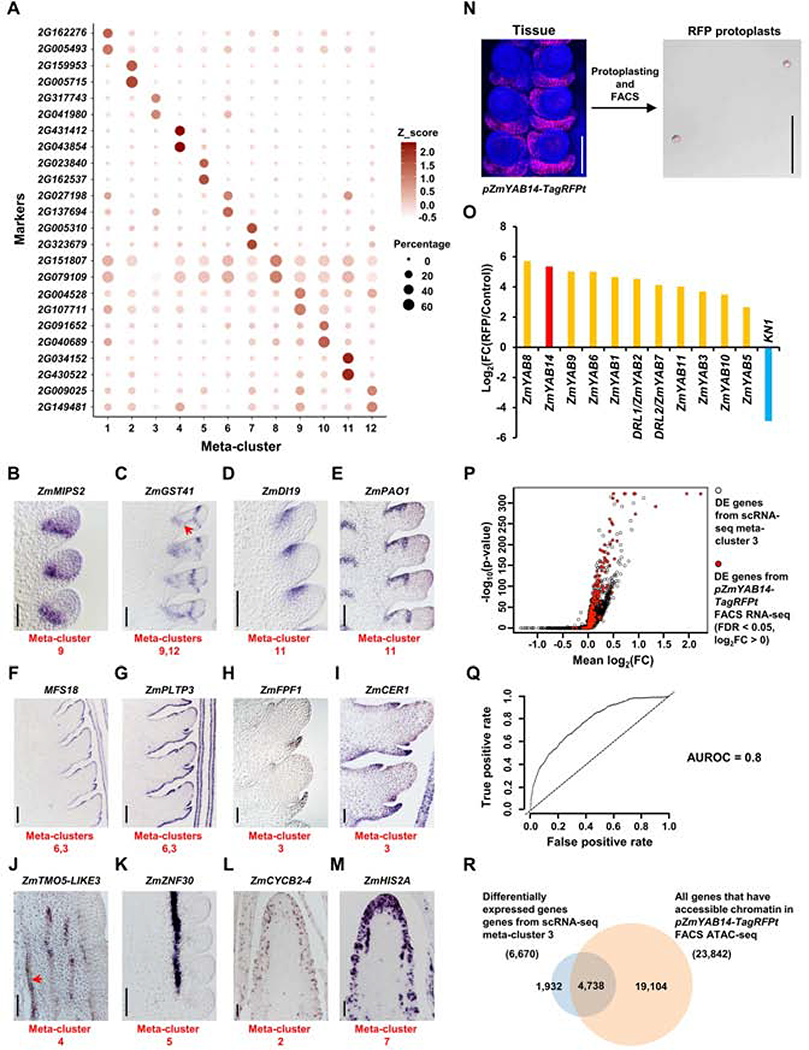
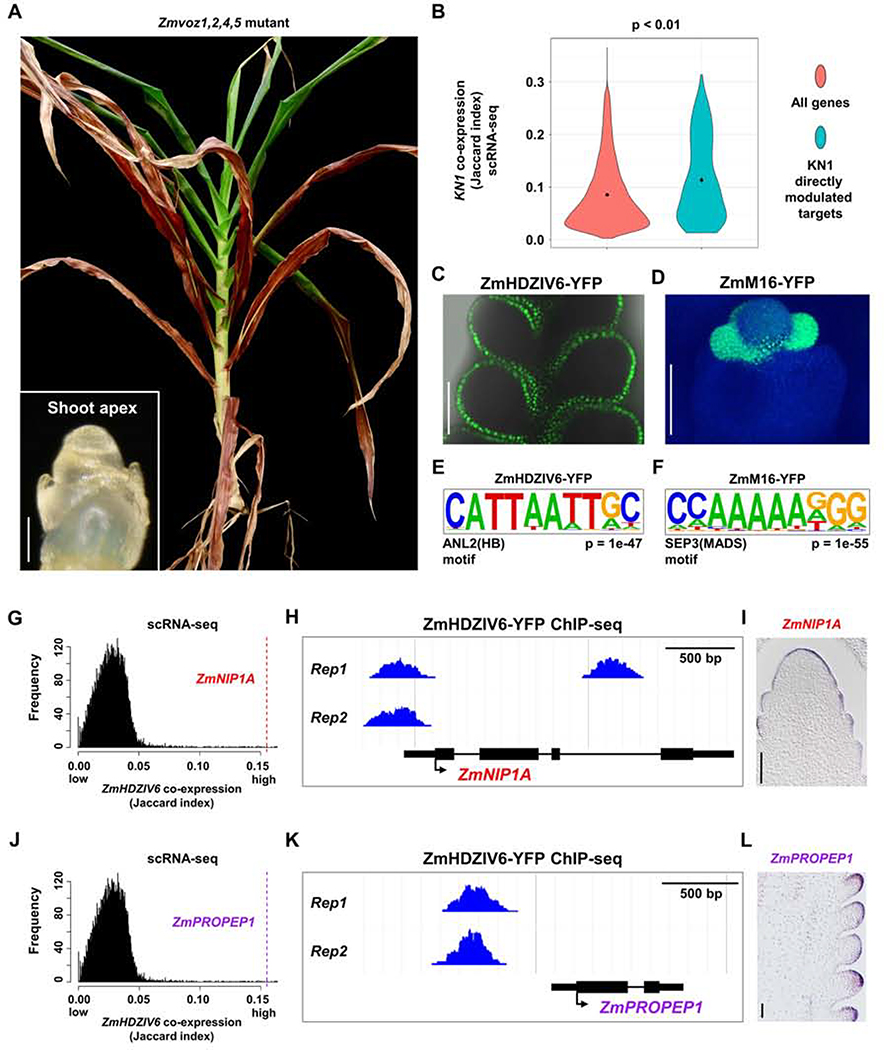
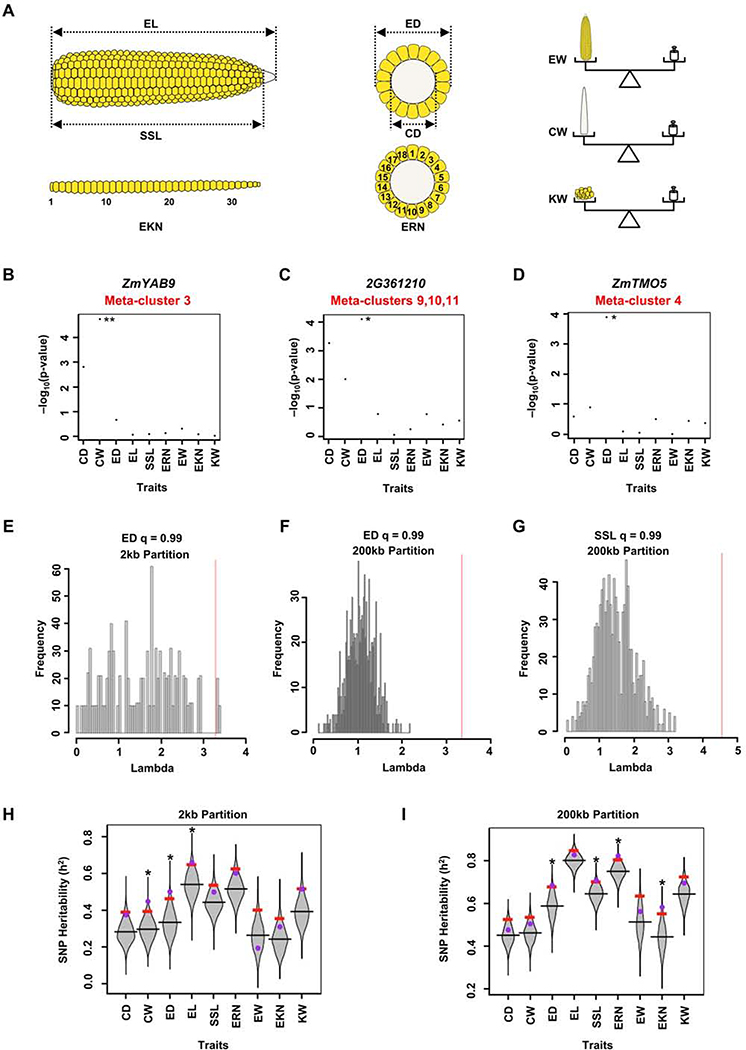
Crop productivity depends on activity of meristems that produce optimized plant architectures, including that of the maize ear. A comprehensive understanding of development requires insight into the full diversity of cell types and developmental domains and the gene networks required to specify them. Until now, these were identified primarily by morphology and insights from classical genetics, which are limited by genetic redundancy and pleiotropy. Here, we investigated the transcriptional profiles of 12,525 single cells from developing maize ears. The resulting developmental atlas provides a single-cell RNA sequencing (scRNA-seq) map of an inflorescence. We validated our results by mRNA in situ hybridization and by fluorescence-activated cell sorting (FACS) RNA-seq, and we show how these data may facilitate genetic studies by predicting genetic redundancy, integrating transcriptional networks, and identifying candidate genes associated with crop yield traits.
Keywords: GWAS; developmental networks; maize ear; meristem; scRNA-seq; trait genes.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Bartlett ME, Williams SK, Taylor Z, DeBlasio S, Goldshmidt A, Hall DH, Schmidt RJ, Jackson DP, and Whipple CJ (2015). The Maize PI/GLO Ortholog Zmm16/sterile tassel silky ear1 Interacts with the Zygomorphy and Sex Determination Pathways in Flower Development. Plant Cell 27, 3081–3098. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
