

Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer
- PMID: 33402734
- PMCID: PMC7785750
- DOI: 10.1038/s41467-020-20430-7
Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer
Abstract
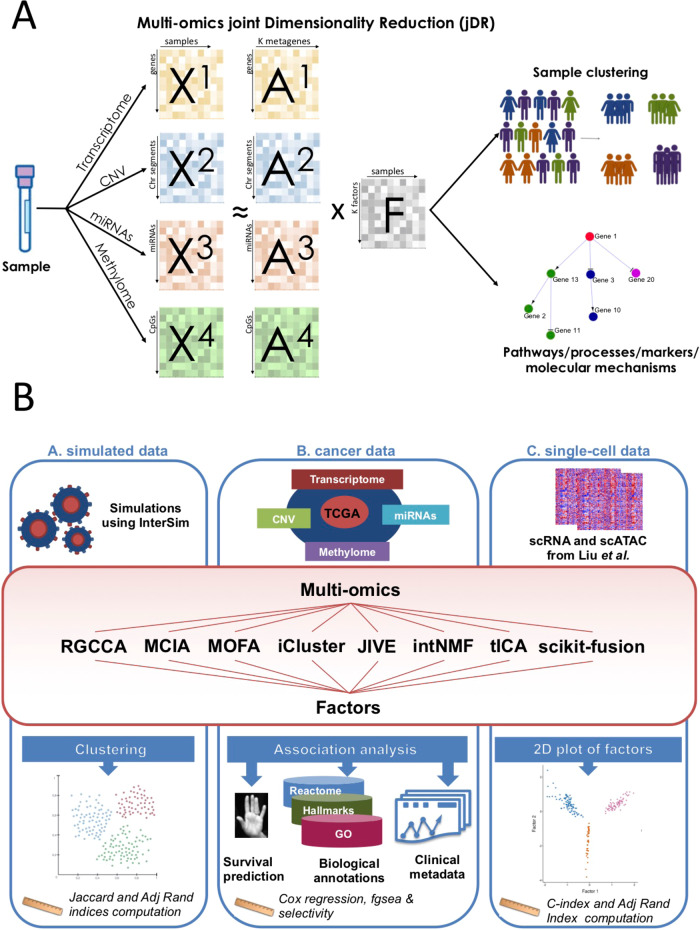
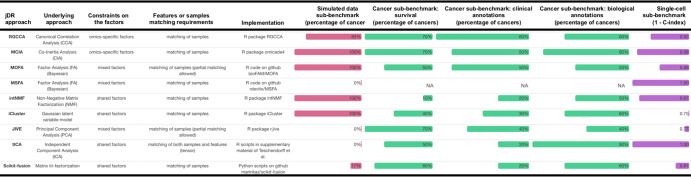
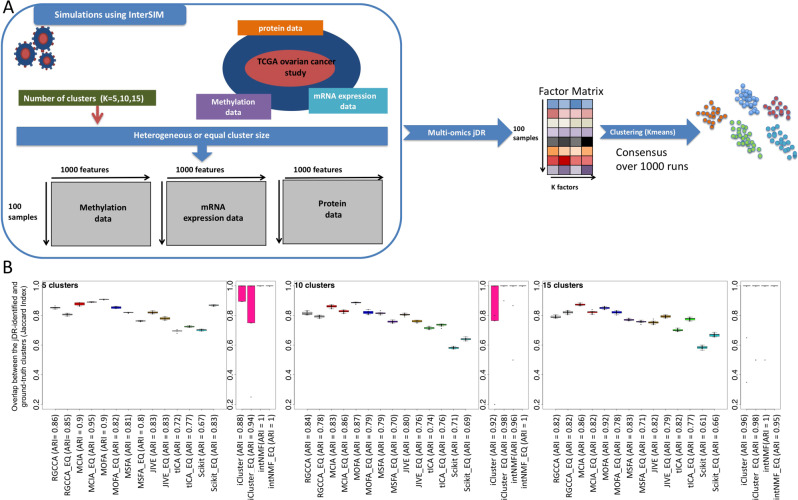
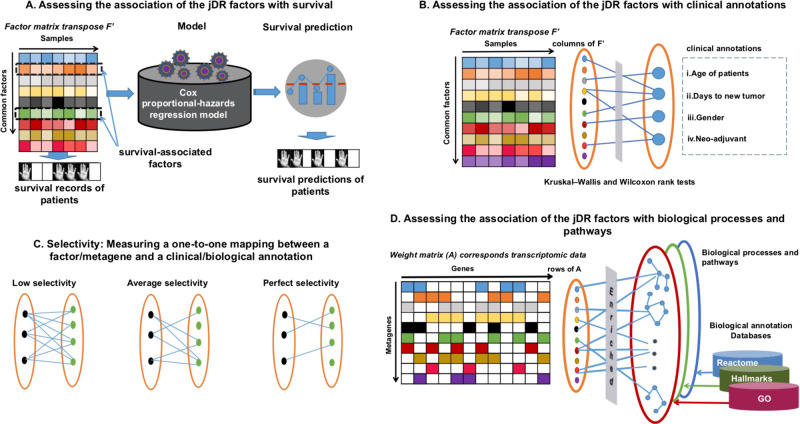
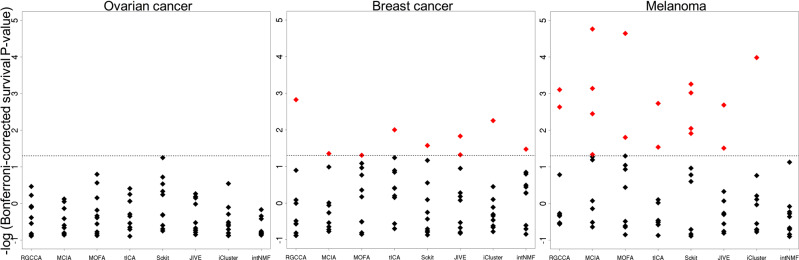
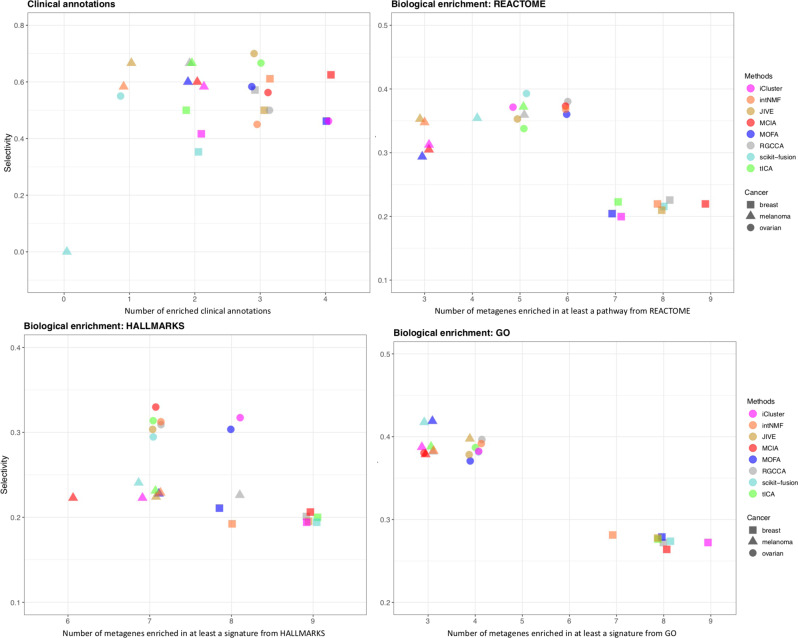
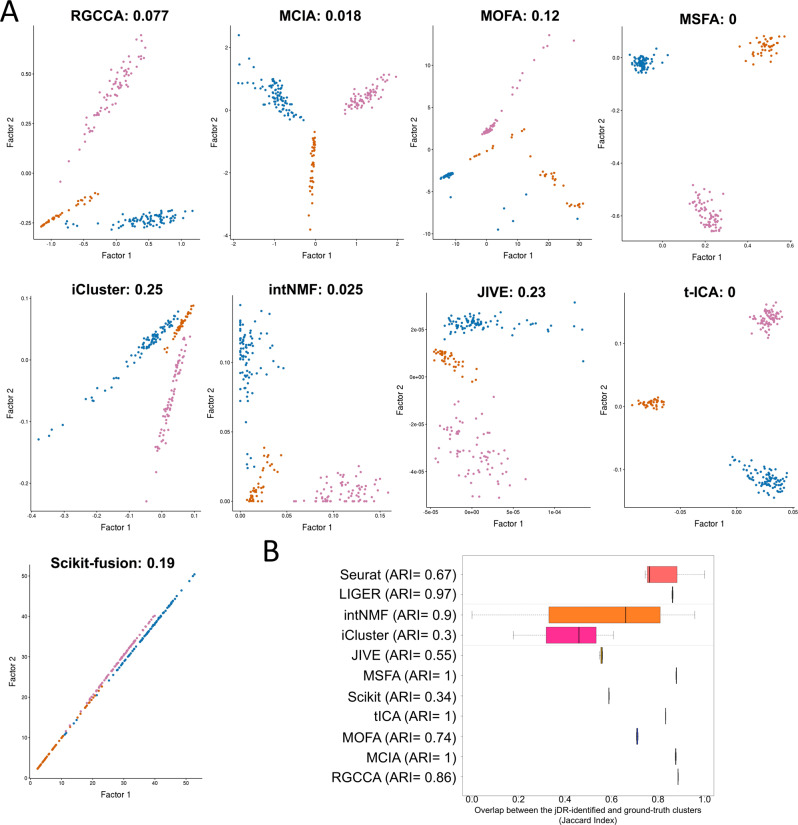
High-dimensional multi-omics data are now standard in biology. They can greatly enhance our understanding of biological systems when effectively integrated. To achieve proper integration, joint Dimensionality Reduction (jDR) methods are among the most efficient approaches. However, several jDR methods are available, urging the need for a comprehensive benchmark with practical guidelines. We perform a systematic evaluation of nine representative jDR methods using three complementary benchmarks. First, we evaluate their performances in retrieving ground-truth sample clustering from simulated multi-omics datasets. Second, we use TCGA cancer data to assess their strengths in predicting survival, clinical annotations and known pathways/biological processes. Finally, we assess their classification of multi-omics single-cell data. From these in-depth comparisons, we observe that intNMF performs best in clustering, while MCIA offers an effective behavior across many contexts. The code developed for this benchmark study is implemented in a Jupyter notebook-multi-omics mix (momix)-to foster reproducibility, and support users and future developers.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
