

The origins and consequences of UPF1 variants in pancreatic adenosquamous carcinoma
- PMID: 33404013
- PMCID: PMC7846273
- DOI: 10.7554/eLife.62209
The origins and consequences of UPF1 variants in pancreatic adenosquamous carcinoma
Abstract
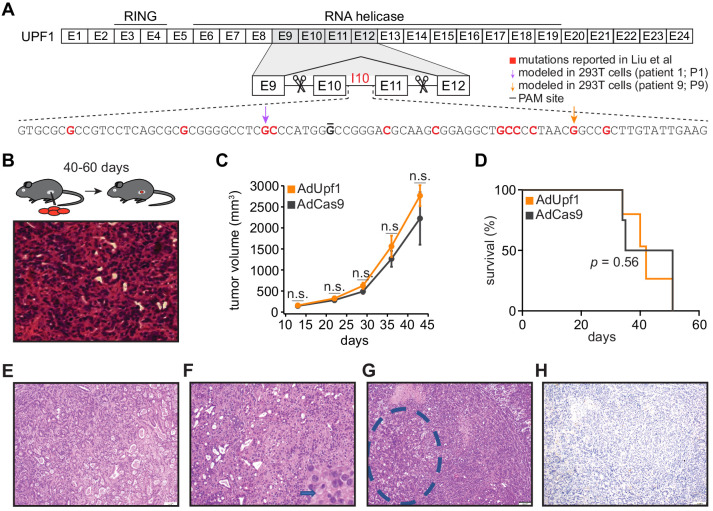
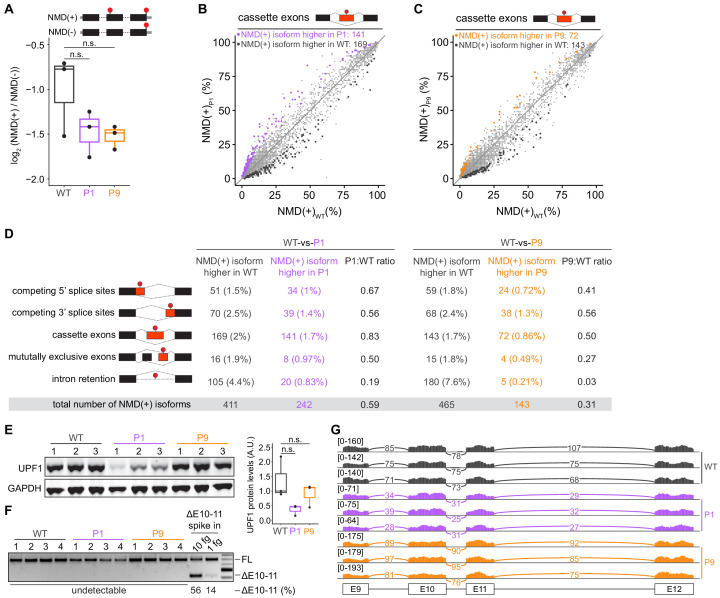
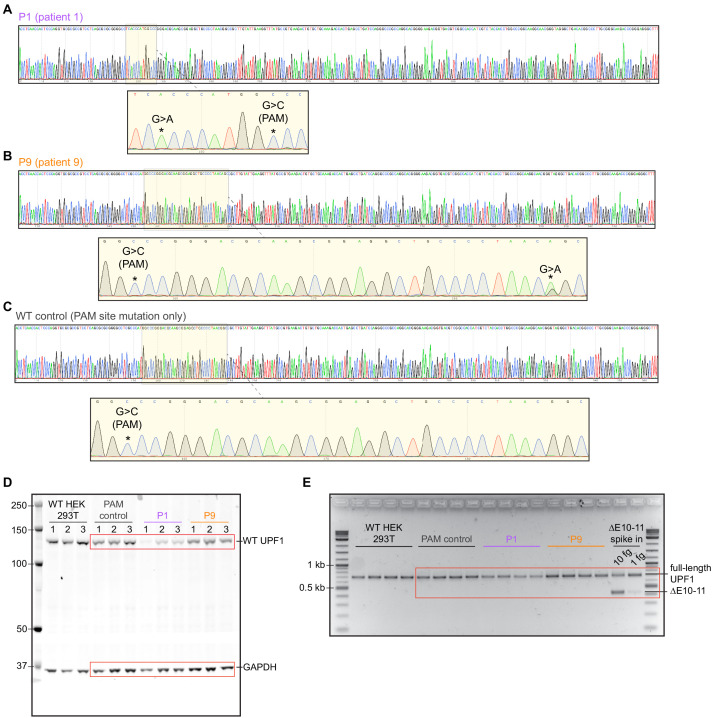
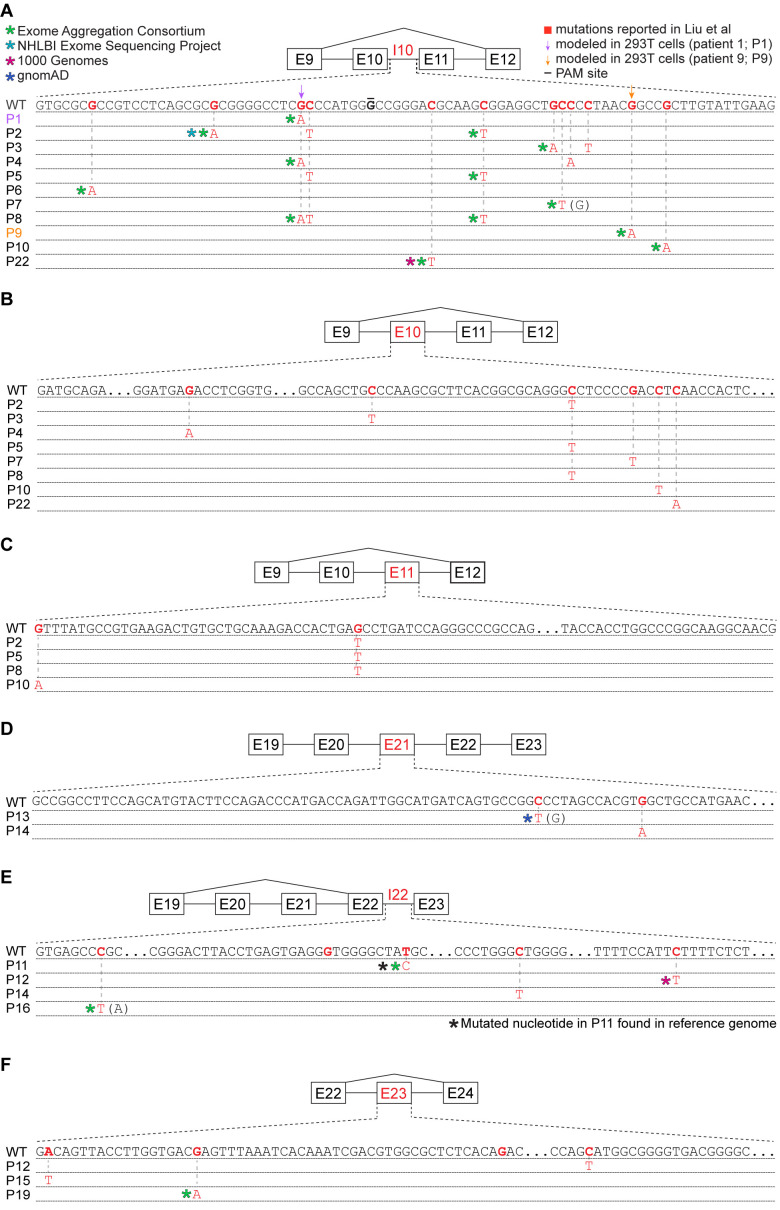
Pancreatic adenosquamous carcinoma (PASC) is an aggressive cancer whose mutational origins are poorly understood. An early study reported high-frequency somatic mutations affecting UPF1, a nonsense-mediated mRNA decay (NMD) factor, in PASC, but subsequent studies did not observe these lesions. The corresponding controversy about whether UPF1 mutations are important contributors to PASC has been exacerbated by a paucity of functional studies. Here, we modeled two UPF1 mutations in human and mouse cells to find no significant effects on pancreatic cancer growth, acquisition of adenosquamous features, UPF1 splicing, UPF1 protein, or NMD efficiency. We subsequently discovered that 45% of UPF1 mutations reportedly present in PASCs are identical to standing genetic variants in the human population, suggesting that they may be non-pathogenic inherited variants rather than pathogenic mutations. Our data suggest that UPF1 is not a common functional driver of PASC and motivate further attempts to understand the genetic origins of these malignancies.
Keywords: cancer biology; cancer genetics; cancer genomics; human; pancreatic cancer.
Plain language summary
Cancer is a group of complex diseases in which cells grow uncontrollably and spread into surrounding tissues and other parts of the body. All types of cancers develop from changes – or mutations – in the genes that affect the pathways involved in controlling the growth of cells. Different cancers possess unique sets of mutations that affect specific genes, and often, it is difficult to determine which of them play the most important role in a particular type of cancer. For example, pancreatic adenosquamous carcinoma, a rare and aggressive form of pancreatic cancer, is a devastating disease with a poor chance of survival – patients rarely live longer than one year after diagnosis. While the cells of this particular cancer display distinct features that separate them from other forms of pancreatic cancer, the genetic causes of these features are unclear. Using new technologies, some researchers have reported mutations in a ‘quality control’ gene called ‘UPF1’, which is responsible for destroying faulty forms of genetic material. However, subsequent studies did not find such mutations. To clarify the role of UPF1 in pancreatic adenosquamous carcinoma, Polaski et al. used mouse and human cancer cells with UPF1 mutations and monitored their effects on tumour growth and the development of features unique to this disease. Polaski et al. first injected mice with mouse pancreatic cancer cells containing mutations in UPF1 (mutated cells) and cancer cells without. Both groups of mice developed pancreatic tumours but there was no difference in tumour growth between the mutated and non-mutated cells, and neither cell type displayed distinct features. The researchers then generated human mutated cells, which were also found to lack any specific characteristics. Further analysis showed that the mutations did not stop UPF1 from working, in fact, over 40% of these mutations occurred naturally in humans without causing cancer. This suggests that UPF1 does not seem to be involved in pancreatic adenosquamous carcinoma. Further investigation is needed to illuminate key genetic players in the development of this type of cancer, which will be vital for improving treatments and outcomes for patients suffering from this disease.
© 2021, Polaski et al.
Conflict of interest statement
JP, DU, LE, GA, SL, AV, RK, RB No competing interests declared
Figures
References
-
- Borazanci E, Millis SZ, Korn R, Han H, Whatcott CJ, Gatalica Z, Barrett MT, Cridebring D, Von Hoff DD. Adenosquamous carcinoma of the pancreas: molecular characterization of 23 patients along with a literature review. World Journal of Gastrointestinal Oncology. 2015;7:132–140. doi: 10.4251/wjgo.v7.i9.132. - DOI - PMC - PubMed
-
- Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, Apken LH, Hobbs GA, Luo R, Lecomte N, Babu S, Pan FC, Alonso-Curbelo D, Morris JP, Askan G, Grbovic-Huezo O, Ogrodowski P, Bermeo J, Saglimbeni J, Cruz CD, Ho YJ, Lawrence SA, Melchor JP, Goda GA, Bai K, Pastore A, Hogg SJ, Raghavan S, Bailey P, Chang DK, Biankin A, Shroyer KR, Wolpin BM, Aguirre AJ, Ventura A, Taylor B, Der CJ, Dominguez D, Kümmel D, Oeckinghaus A, Lowe SW, Bradley RK, Abdel-Wahab O, Leach SD. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic Cancer. Cancer Cell. 2020;38:198–211. doi: 10.1016/j.ccell.2020.05.010. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
