

The role of calcium homeostasis remodeling in inherited cardiac arrhythmia syndromes
- PMID: 33404893
- PMCID: PMC7940310
- DOI: 10.1007/s00424-020-02505-y
The role of calcium homeostasis remodeling in inherited cardiac arrhythmia syndromes
Abstract
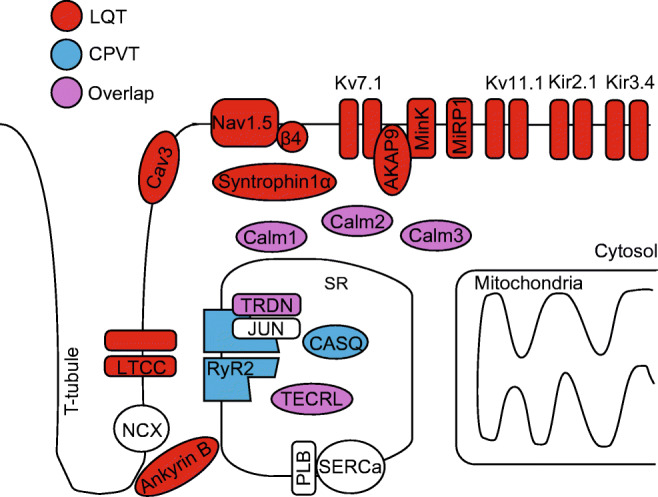
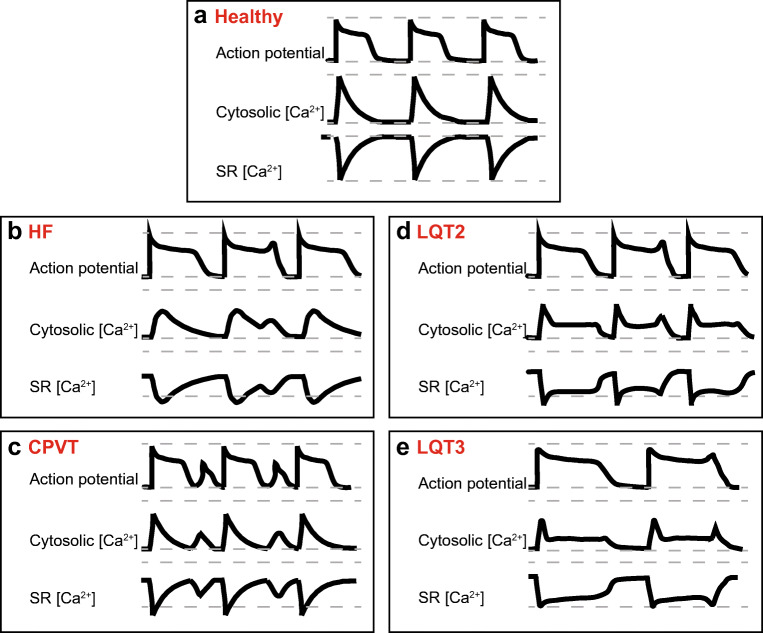
Sudden cardiac death due to malignant ventricular arrhythmias remains the major cause of mortality in the postindustrial world. Defective intracellular Ca2+ homeostasis has been well established as a key contributing factor to the enhanced propensity for arrhythmia in acquired cardiac disease, such as heart failure or diabetic cardiomyopathy. More recent advances provide a strong basis to the emerging view that hereditary cardiac arrhythmia syndromes are accompanied by maladaptive remodeling of Ca2+ homeostasis which substantially increases arrhythmic risk. This brief review will focus on functional changes in elements of Ca2+ handling machinery in cardiomyocytes that occur secondary to genetic mutations associated with catecholaminergic polymorphic ventricular tachycardia, and long QT syndrome.
Keywords: Calcium homeostasis remodeling; Calcium-dependent arrhythmia; Catecholaminergic polymorphic ventricular tachycardia; Heart failure; Long QT syndrome.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Altmann HM, Tester DJ, Will ML, Middha S, Evans JM, Eckloff BW, Ackerman MJ. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: elucidation of the triadin knockout syndrome. Circulation. 2015;131(23):2051–2060. doi: 10.1161/CIRCULATIONAHA.115.015397. - DOI - PubMed
-
- Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6(12):e28324. doi: 10.1371/journal.pone.0028324. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
