

mixIndependR: a R package for statistical independence testing of loci in database of multi-locus genotypes
- PMID: 33407074
- PMCID: PMC7788837
- DOI: 10.1186/s12859-020-03945-0
mixIndependR: a R package for statistical independence testing of loci in database of multi-locus genotypes
Abstract
Background: Multi-locus genotype data are widely used in population genetics and disease studies. In evaluating the utility of multi-locus data, the independence of markers is commonly considered in many genomic assessments. Generally, pairwise non-random associations are tested by linkage disequilibrium; however, the dependence of one panel might be triplet, quartet, or other. Therefore, a compatible and user-friendly software is necessary for testing and assessing the global linkage disequilibrium among mixed genetic data.
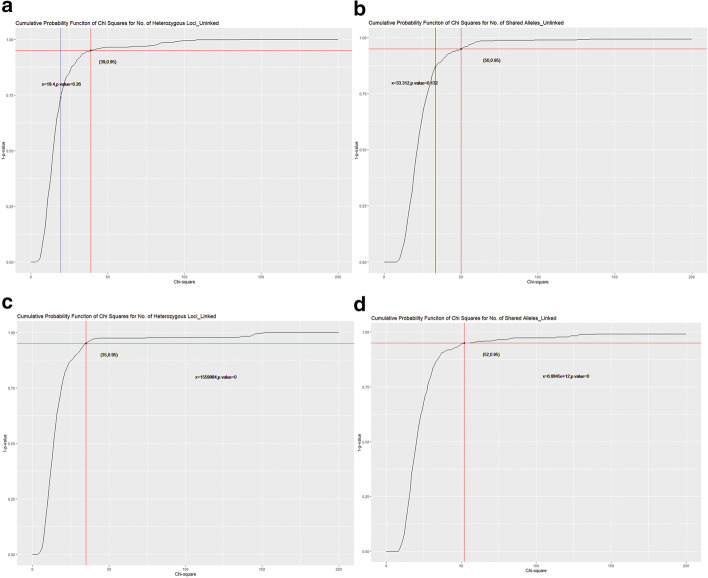
Results: This study describes a software package for testing the mutual independence of mixed genetic datasets. Mutual independence is defined as no non-random associations among all subsets of the tested panel. The new R package "mixIndependR" calculates basic genetic parameters like allele frequency, genotype frequency, heterozygosity, Hardy-Weinberg equilibrium, and linkage disequilibrium (LD) by mutual independence from population data, regardless of the type of markers, such as simple nucleotide polymorphisms, short tandem repeats, insertions and deletions, and any other genetic markers. A novel method of assessing the dependence of mixed genetic panels is developed in this study and functionally analyzed in the software package. By comparing the observed distribution of two common summary statistics (the number of heterozygous loci [K] and the number of share alleles [X]) with their expected distributions under the assumption of mutual independence, the overall independence is tested.
Conclusion: The package "mixIndependR" is compatible to all categories of genetic markers and detects the overall non-random associations. Compared to pairwise disequilibrium, the approach described herein tends to have higher power, especially when number of markers is large. With this package, more multi-functional or stronger genetic panels can be developed, like mixed panels with different kinds of markers. In population genetics, the package "mixIndependR" makes it possible to discover more about admixture of populations, natural selection, genetic drift, and population demographics, as a more powerful method of detecting LD. Moreover, this new approach can optimize variants selection in disease studies and contribute to panel combination for treatments in multimorbidity. Application of this approach in real data is expected in the future, and this might bring a leap in the field of genetic technology.
Availability: The R package mixIndependR, is available on the Comprehensive R Archive Network (CRAN) at: https://cran.r-project.org/web/packages/mixIndependR/index.html .
Keywords: Linkage disequilibrium; Mutual independence; Non-random association; R package; SNPs; STRs.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
-
- Butler JM, Coble MD, Vallone PM. STRs vs. SNPs: thoughts on the future of forensic DNA testing. Forensic Sci Med Pathol. 2007;3(3):200–205. - PubMed
-
- Wang L, He W, Mao J, Wang H, Jin B, Luo HB, Liang WB, Zhang L. Development of a SNP-STRs multiplex for forensic identification. Forensic Sci Int Genet Suppl Ser. 2015;5:e598–e600.
-
- Schulze TG, Chen YS, Akula N, Hennessy K, Badner JA, McInnis MG, DePaulo JR, Schumacher J, Cichon S, Propping P, et al. Can long-range microsatellite data be used to predict short-range linkage disequilibrium? Hum Mol Genet. 2002;11(12):1363–1372. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
