

Infantile onset Sandhoff disease: clinical manifestation and a novel common mutation in Thai patients
- PMID: 33407268
- PMCID: PMC7789739
- DOI: 10.1186/s12887-020-02481-3
Infantile onset Sandhoff disease: clinical manifestation and a novel common mutation in Thai patients
Abstract
Background: Sandhoff disease (SD) is an autosomal recessive lysosomal storage disorder, resulting in accumulation of GM2 ganglioside, particular in neuronal cells. The disorder is caused by deficiency of β-hexosaminidase B (HEX-B), due to pathogenic variant of human HEXB gene.
Method: This study describes clinical features, biochemical, and genetic defects among Thai patients with infantile SD during 2008-2019.
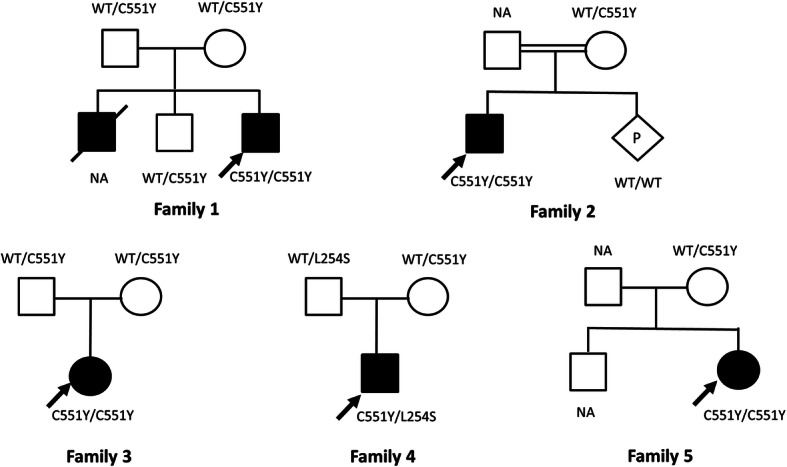
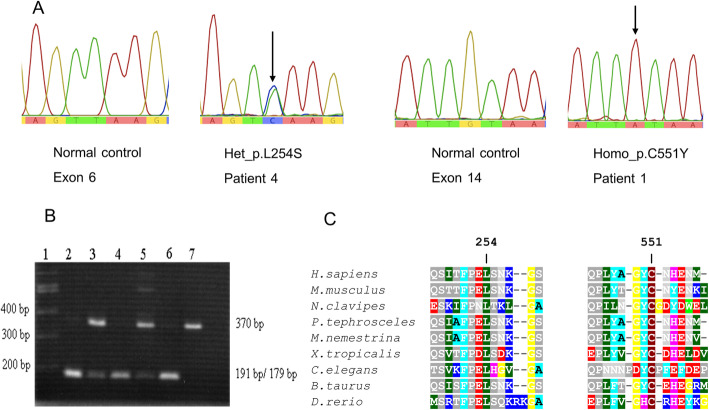
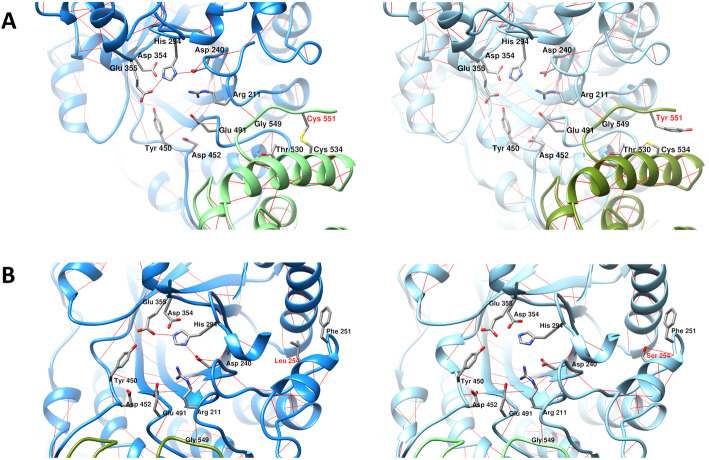
Results: Five unrelated Thai patients presenting with developmental regression, axial hypotonia, seizures, exaggerated startle response to noise, and macular cherry red spot were confirmed to have infantile SD based on deficient HEX enzyme activities and biallelic variants of the HEXB gene. In addition, an uncommon presenting feature, cardiac defect, was observed in one patient. All the patients died in their early childhood. Plasma total HEX and HEX-B activities were severely deficient. Sequencing analysis of HEXB gene identified two variants including c.1652G>A (p.Cys551Tyr) and a novel variant of c.761T>C (p.Leu254Ser), in 90 and 10% of the mutant alleles found, respectively. The results from in silico analysis using multiple bioinformatics tools were in agreement that the p.Cys551Tyr and the p.Leu254Ser are likely pathogenic variants. Molecular modelling suggested that the Cys551Tyr disrupt disulfide bond, leading to protein destabilization while the Leu254Ser resulted in change of secondary structure from helix to coil and disturbing conformation of the active site of the enzyme. Genome-wide SNP array analysis showed no significant relatedness between the five affected individuals. These two variants were not present in control individuals. The prevalence of infantile SD in Thai population is estimated 1 in 1,458,521 and carrier frequency at 1 in 604.
Conclusion: The study suggests that SD likely represents the most common subtype of rare infantile GM2 gangliosidosis identified among Thai patients. We firstly described a potential common variant in HEXB in Thai patients with infantile onset SD. The data can aid a rapid molecular confirmation of infantile SD starting with the hotspot variant and the use of expanded carrier testing.
Keywords: Developmental regression; GM2 gangliosidosis; HEXB; Neurometabolic disorder; Sandhoff disease; Tay-Sachs disease; Thai.
Conflict of interest statement
All authors declare that they have no conflict of interests.
Figures
References
-
- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455(2–3):105–138. - PubMed
-
- Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54(2):176–182. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
