The influence of evolutionary history on human health and disease
- PMID: 33408383
- PMCID: PMC7787134
- DOI: 10.1038/s41576-020-00305-9
The influence of evolutionary history on human health and disease
Abstract
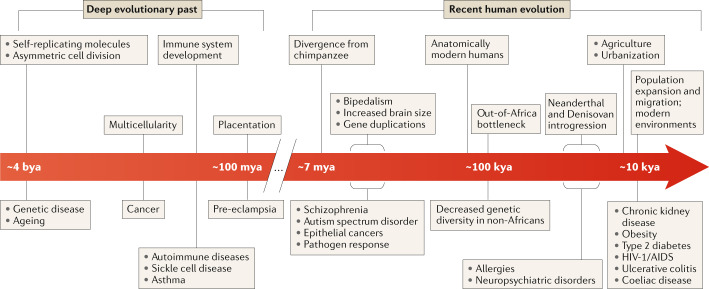
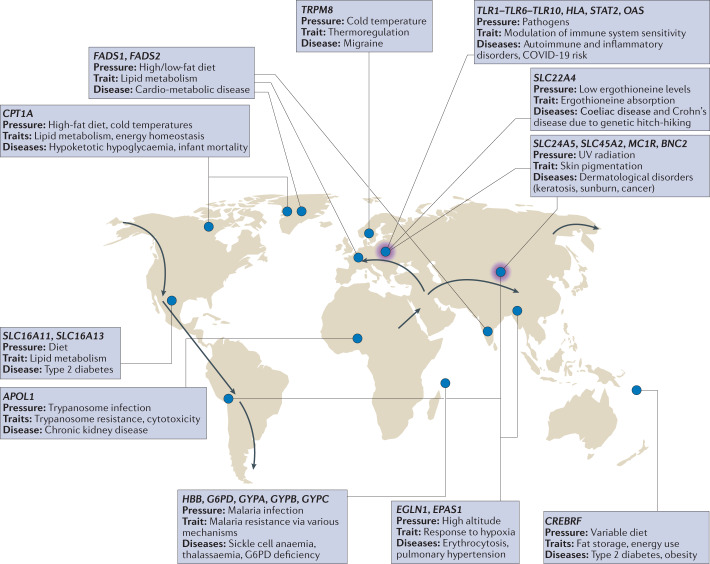
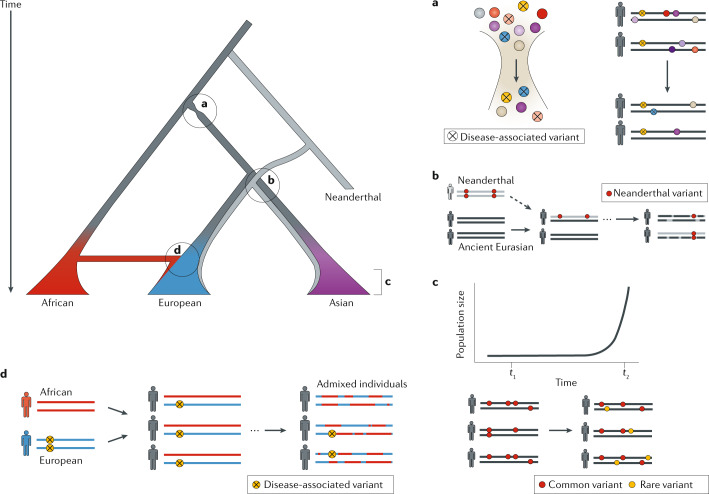
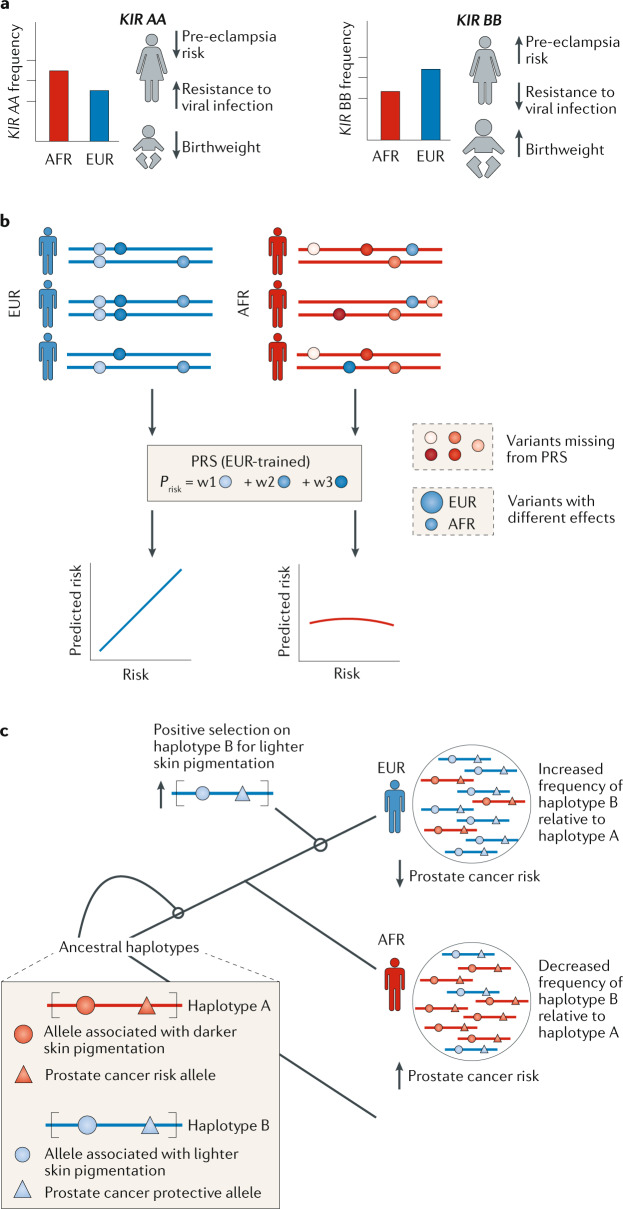
Nearly all genetic variants that influence disease risk have human-specific origins; however, the systems they influence have ancient roots that often trace back to evolutionary events long before the origin of humans. Here, we review how advances in our understanding of the genetic architectures of diseases, recent human evolution and deep evolutionary history can help explain how and why humans in modern environments become ill. Human populations exhibit differences in the prevalence of many common and rare genetic diseases. These differences are largely the result of the diverse environmental, cultural, demographic and genetic histories of modern human populations. Synthesizing our growing knowledge of evolutionary history with genetic medicine, while accounting for environmental and social factors, will help to achieve the promise of personalized genomics and realize the potential hidden in an individual's DNA sequence to guide clinical decisions. In short, precision medicine is fundamentally evolutionary medicine, and integration of evolutionary perspectives into the clinic will support the realization of its full potential.
Conflict of interest statement
The authors declare no competing interests.
Figures