

In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice
- PMID: 33408413
- PMCID: PMC7872200
- DOI: 10.1038/s41586-020-03086-7
In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice
Abstract
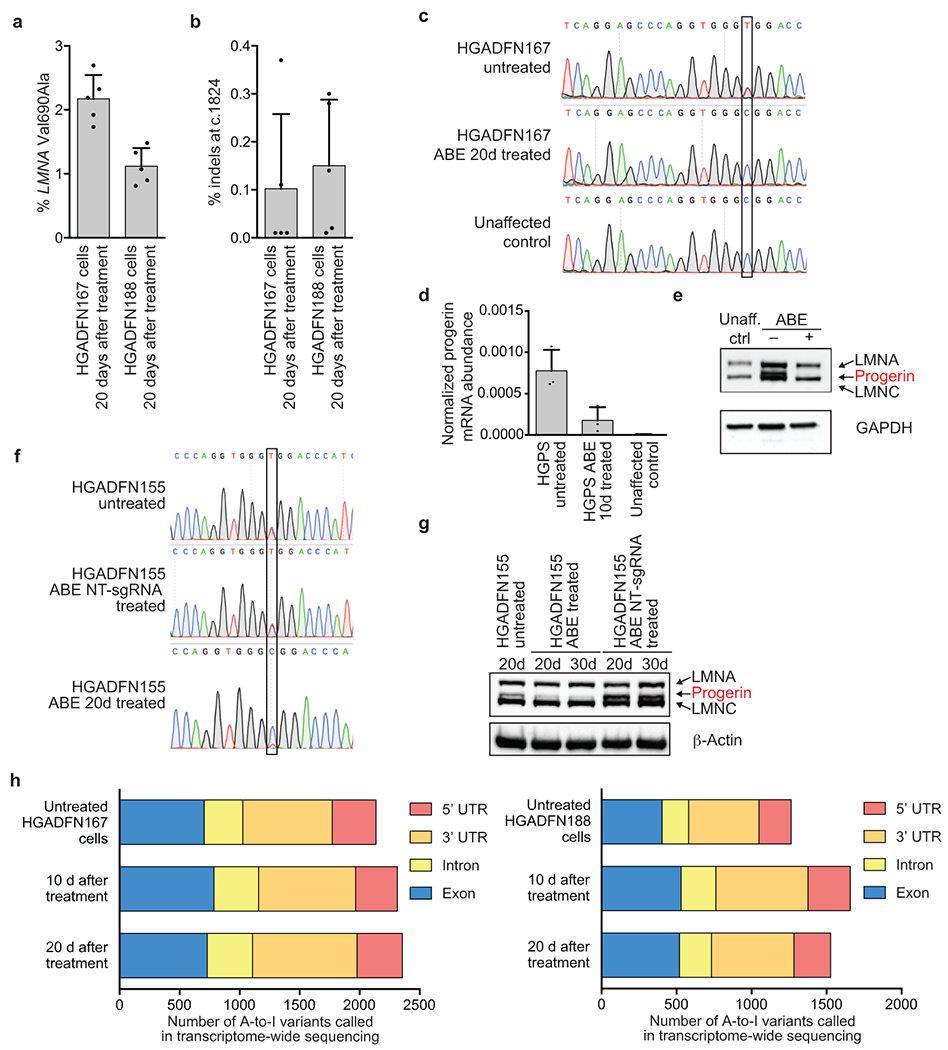
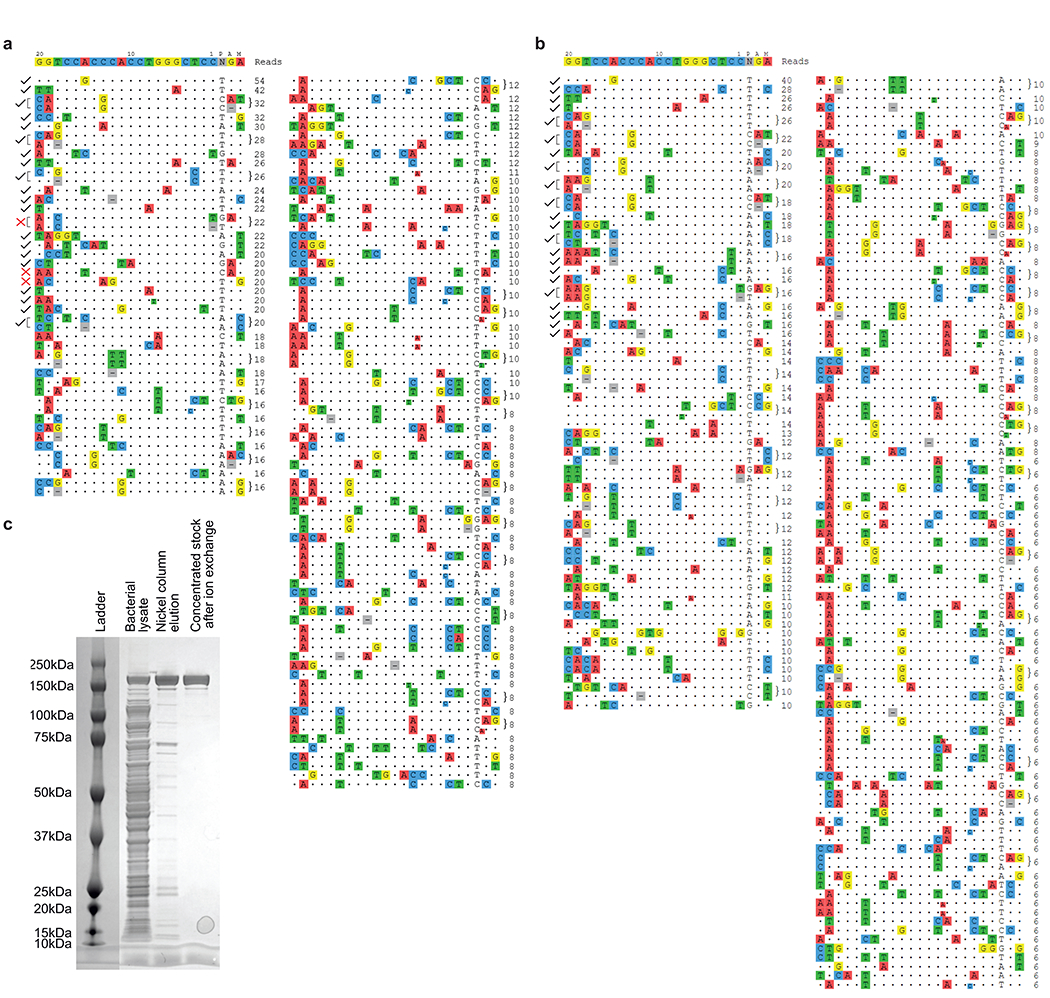
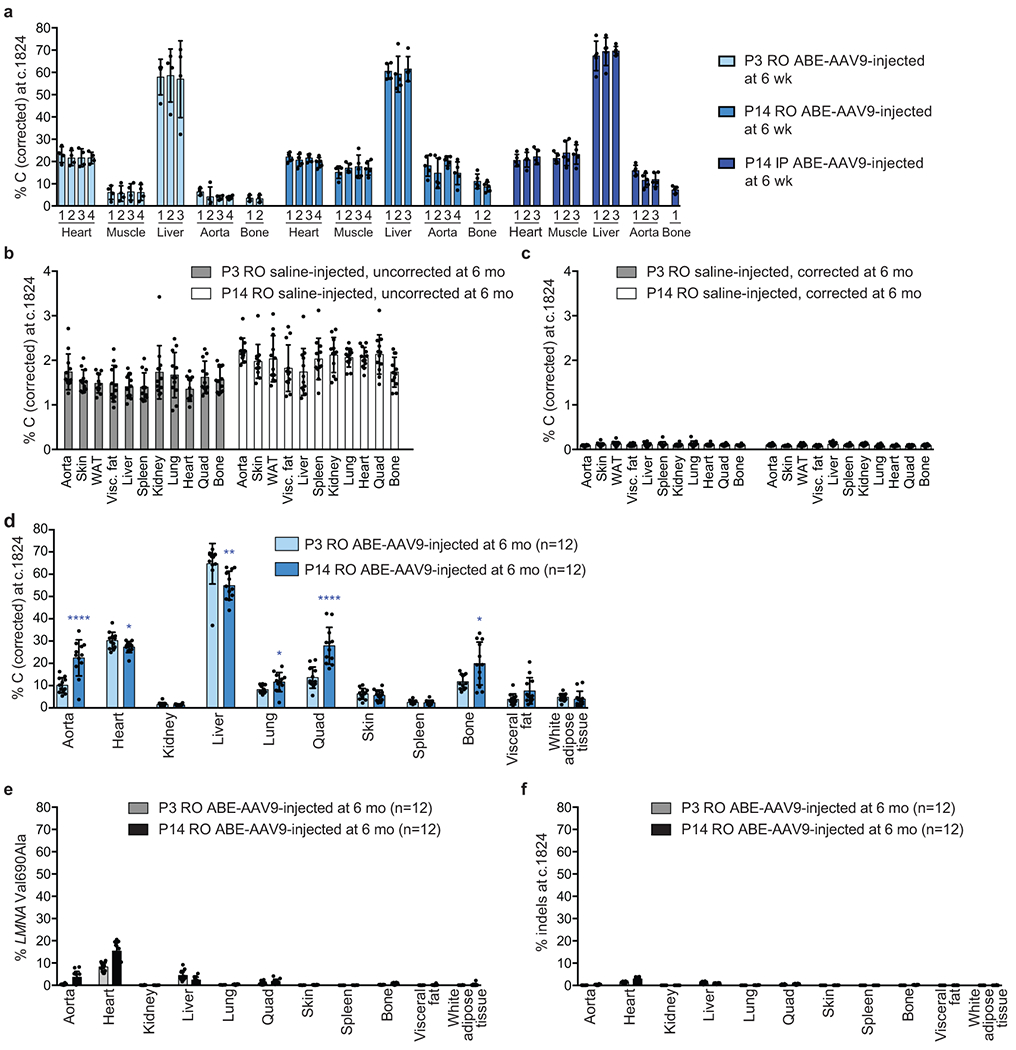
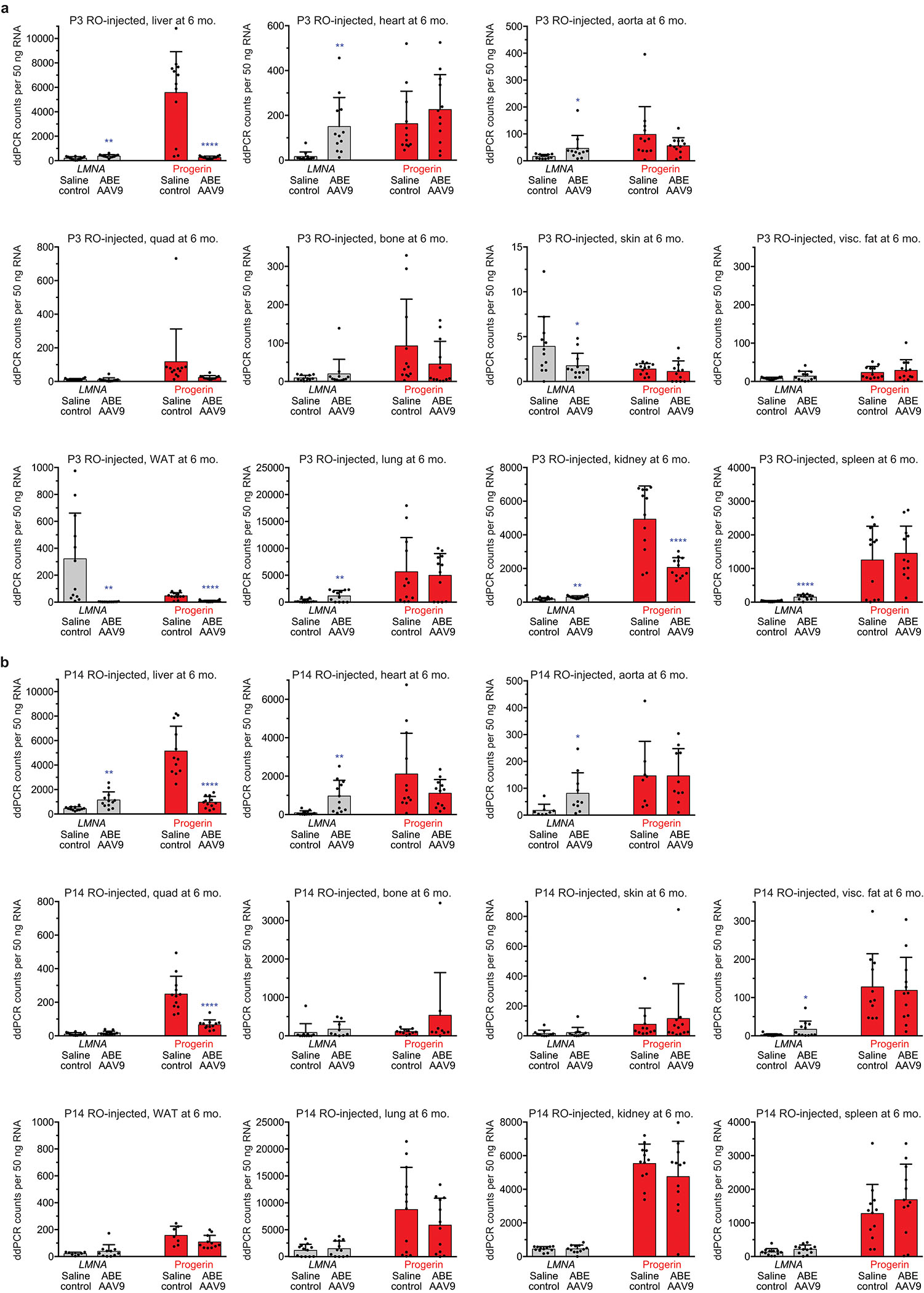
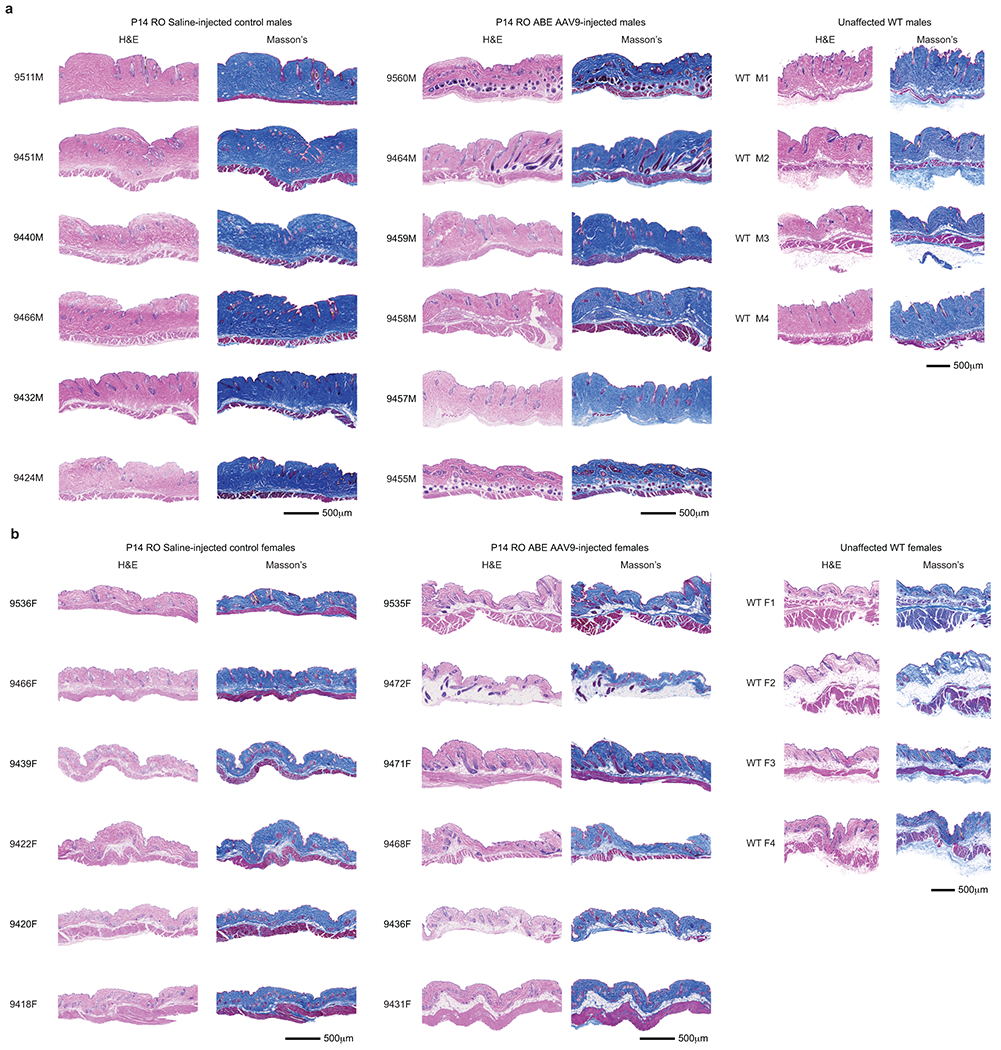
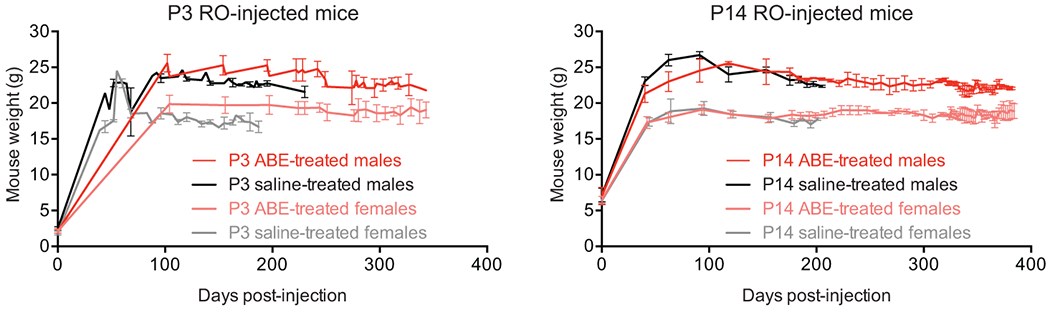
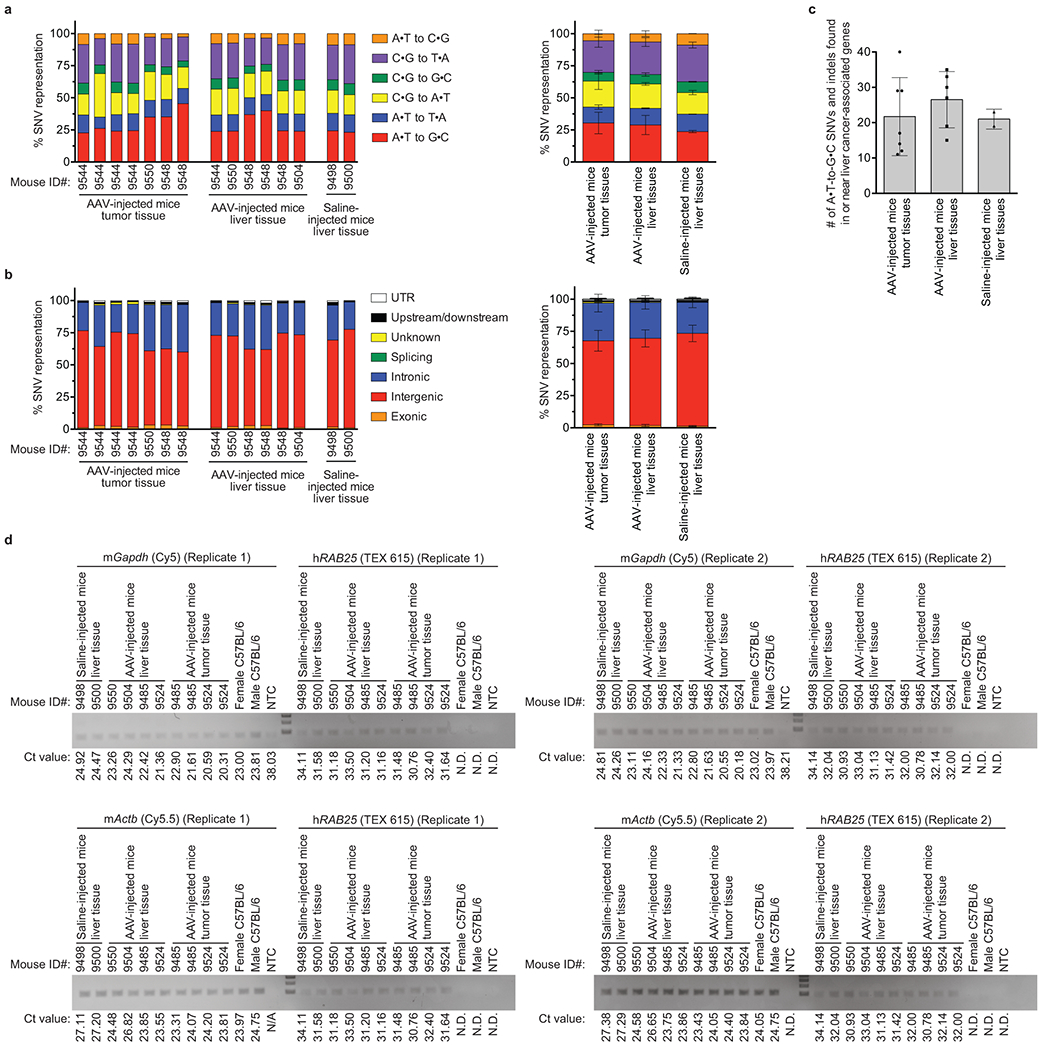
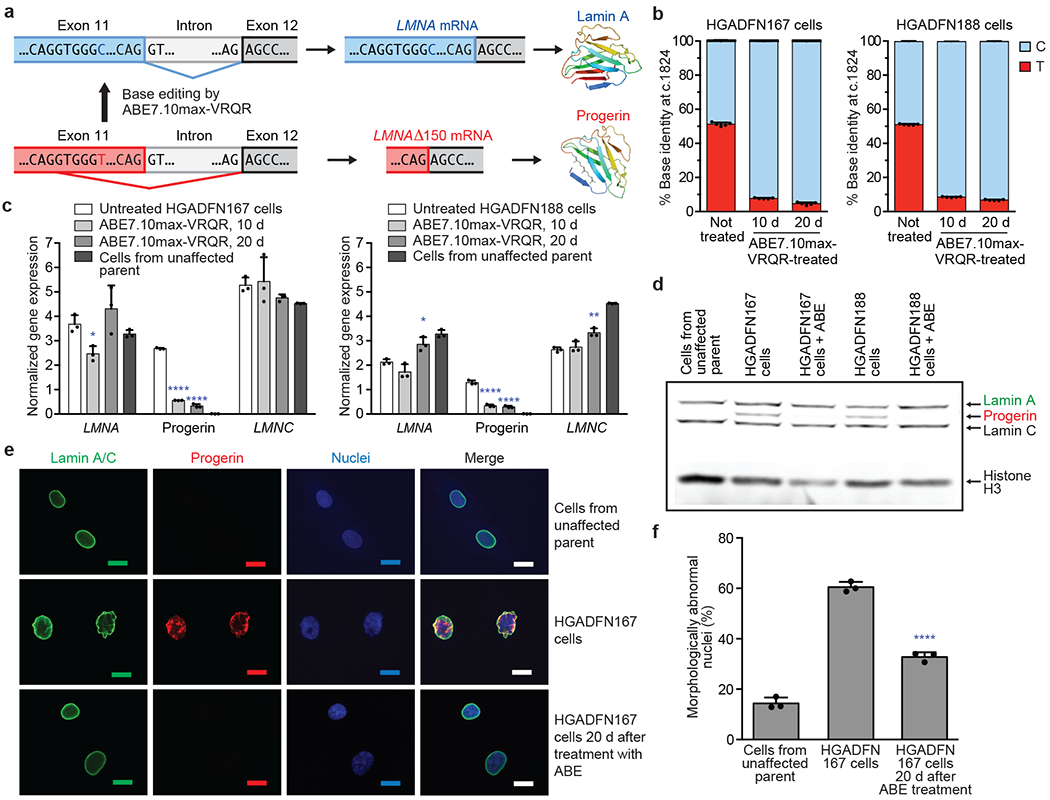
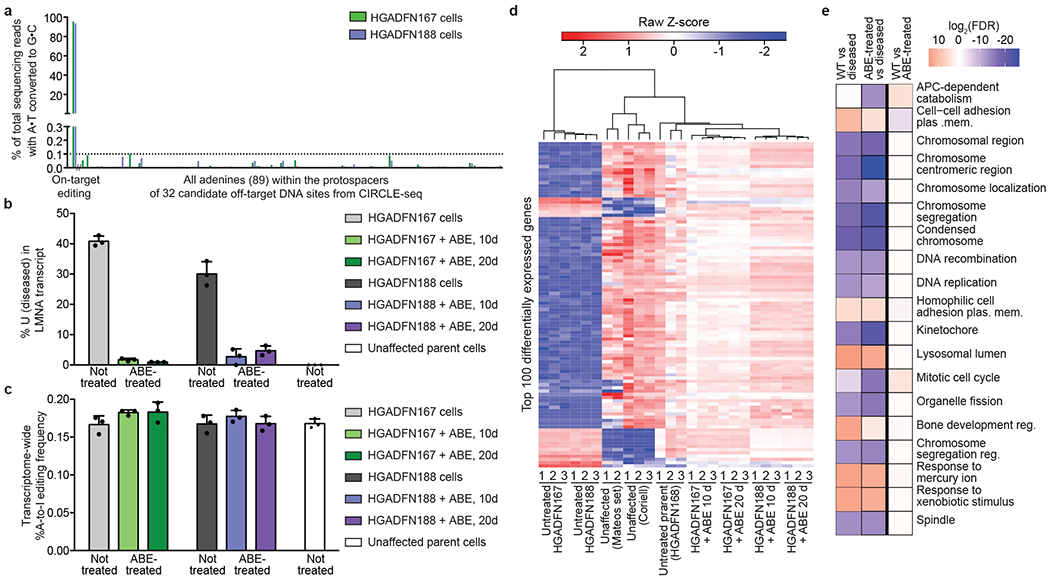
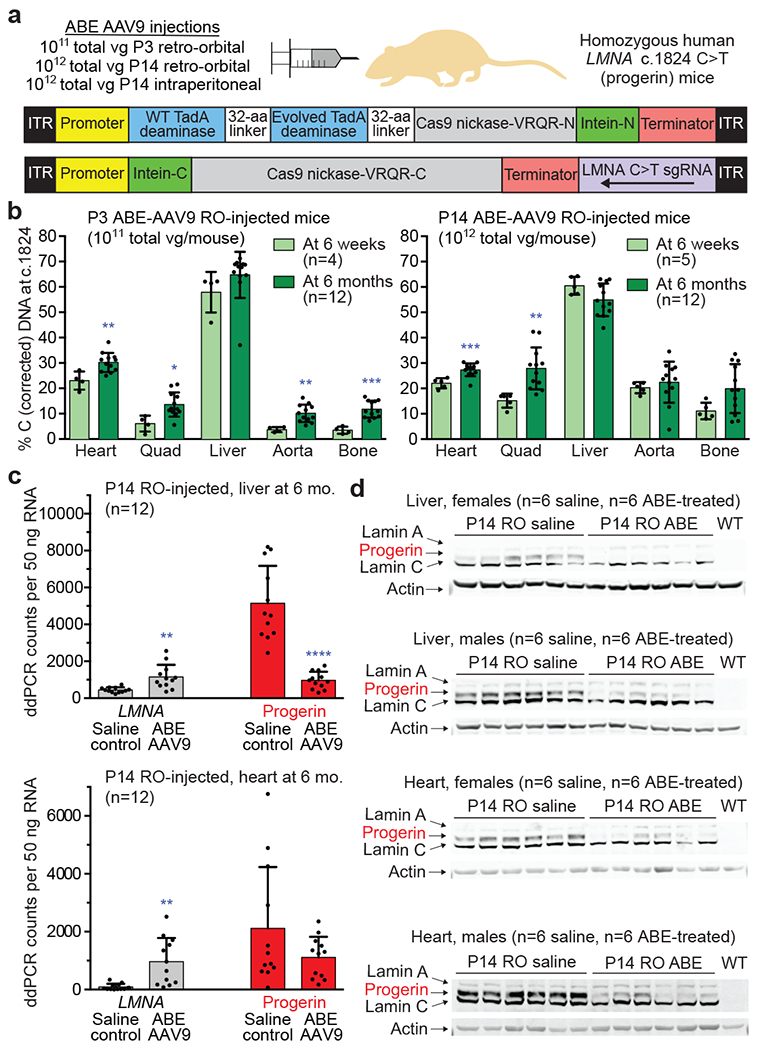
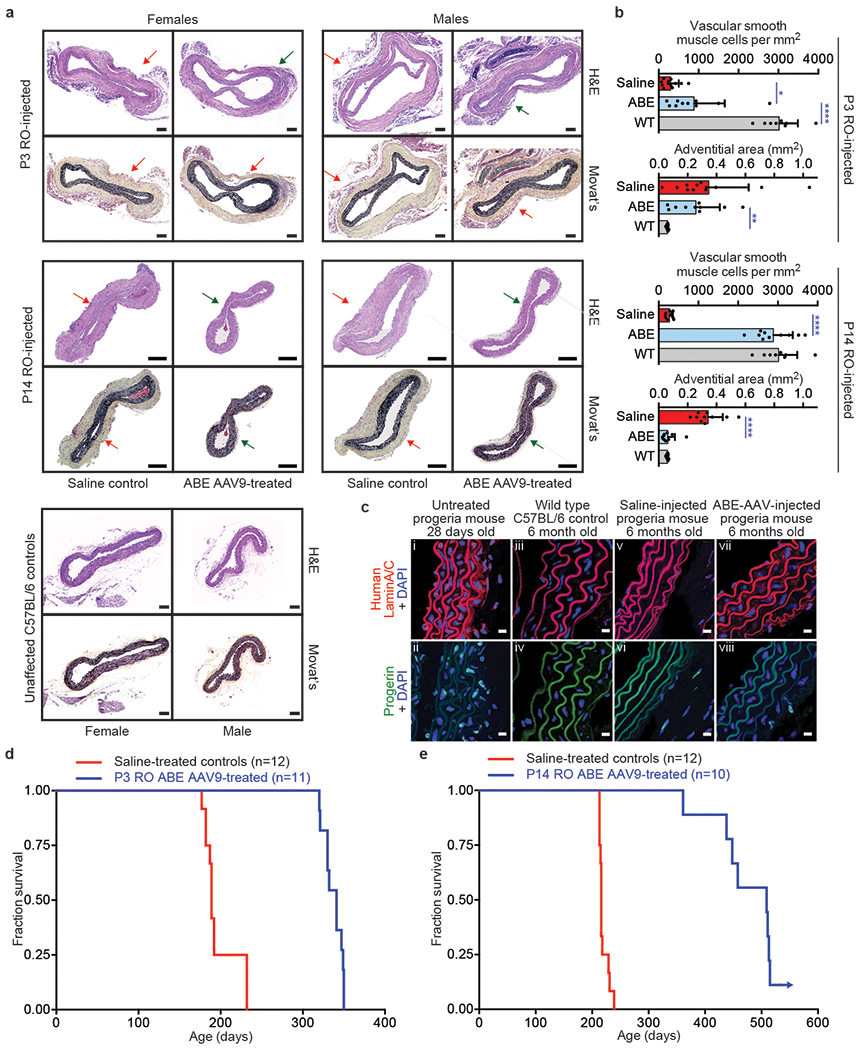
Hutchinson-Gilford progeria syndrome (HGPS or progeria) is typically caused by a dominant-negative C•G-to-T•A mutation (c.1824 C>T; p.G608G) in LMNA, the gene that encodes nuclear lamin A. This mutation causes RNA mis-splicing that produces progerin, a toxic protein that induces rapid ageing and shortens the lifespan of children with progeria to approximately 14 years1-4. Adenine base editors (ABEs) convert targeted A•T base pairs to G•C base pairs with minimal by-products and without requiring double-strand DNA breaks or donor DNA templates5,6. Here we describe the use of an ABE to directly correct the pathogenic HGPS mutation in cultured fibroblasts derived from children with progeria and in a mouse model of HGPS. Lentiviral delivery of the ABE to fibroblasts from children with HGPS resulted in 87-91% correction of the pathogenic allele, mitigation of RNA mis-splicing, reduced levels of progerin and correction of nuclear abnormalities. Unbiased off-target DNA and RNA editing analysis did not detect off-target editing in treated patient-derived fibroblasts. In transgenic mice that are homozygous for the human LMNA c.1824 C>T allele, a single retro-orbital injection of adeno-associated virus 9 (AAV9) encoding the ABE resulted in substantial, durable correction of the pathogenic mutation (around 20-60% across various organs six months after injection), restoration of normal RNA splicing and reduction of progerin protein levels. In vivo base editing rescued the vascular pathology of the mice, preserving vascular smooth muscle cell counts and preventing adventitial fibrosis. A single injection of ABE-expressing AAV9 at postnatal day 14 improved vitality and greatly extended the median lifespan of the mice from 215 to 510 days. These findings demonstrate the potential of in vivo base editing as a possible treatment for HGPS and other genetic diseases by directly correcting their root cause.
Conflict of interest statement
The authors declare competing financial interests: D.R.L. is a co-founder of Beam Therapeutics, Prime Medicine, Pairwise Plants, and Editas Medicine, companies that use genome editing.
Figures
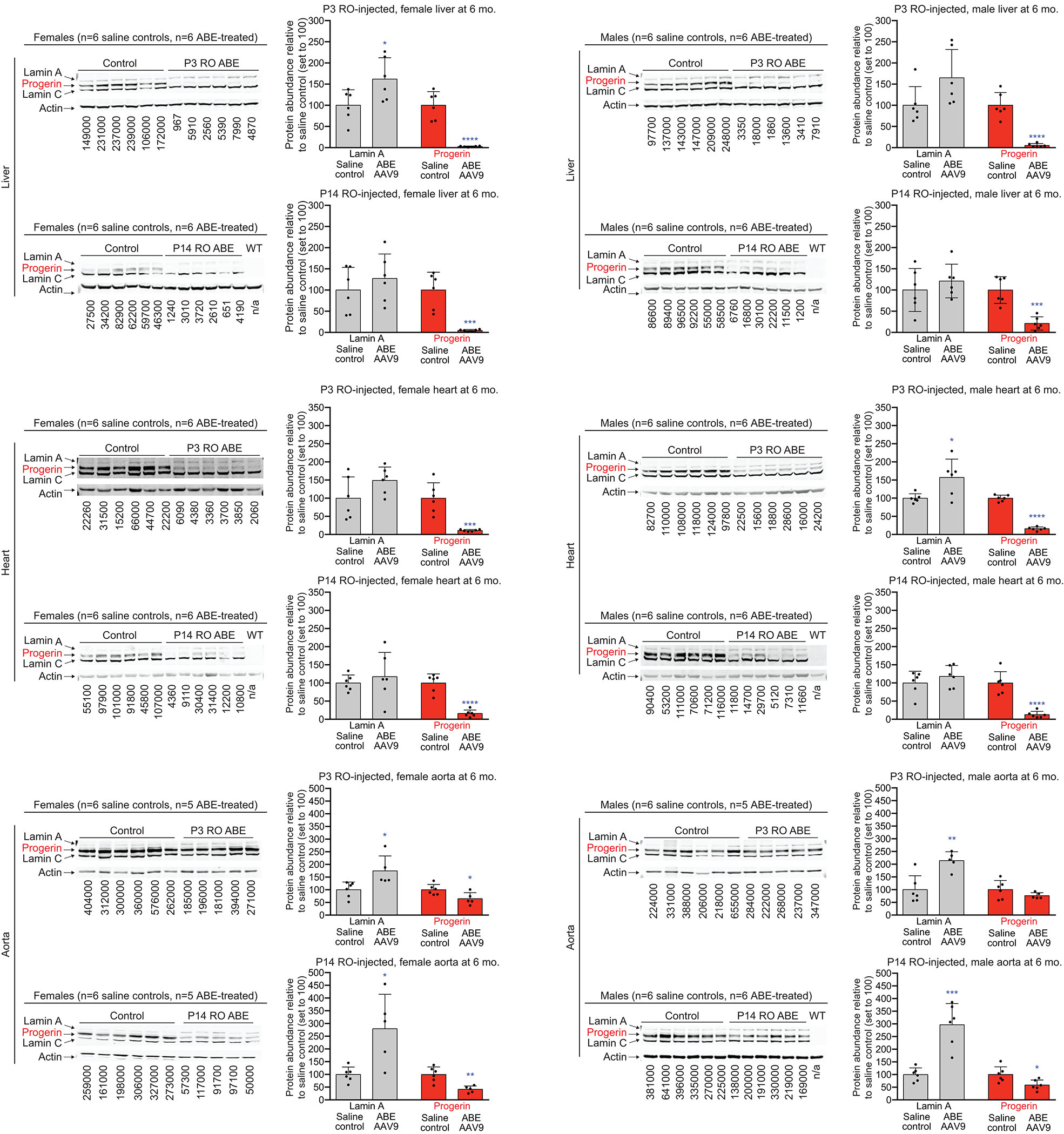
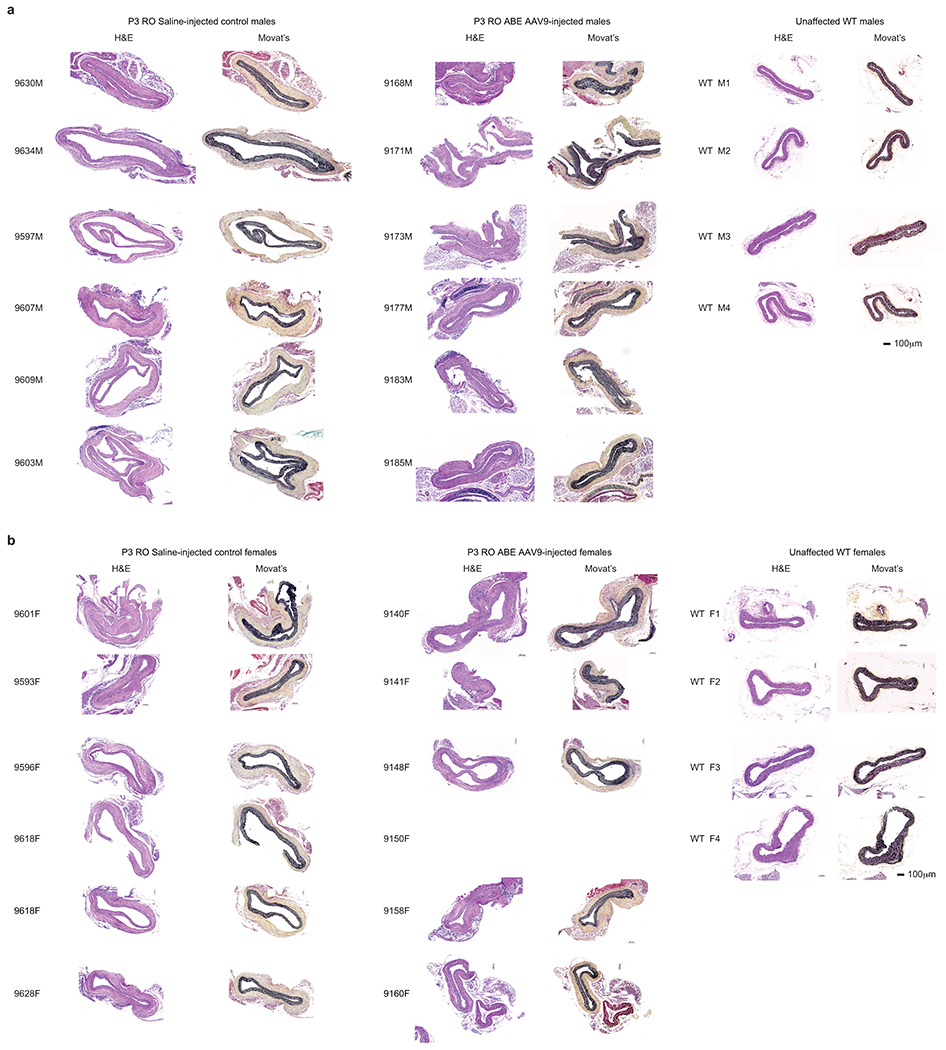
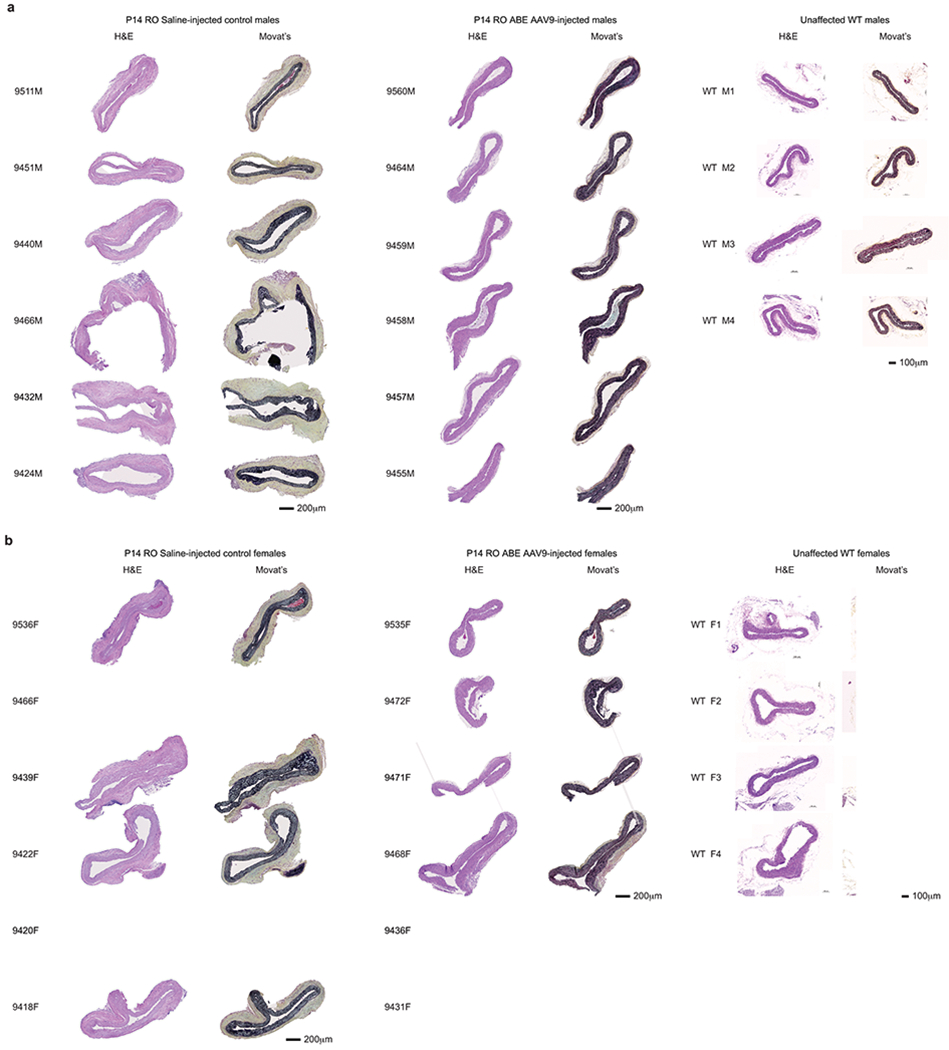
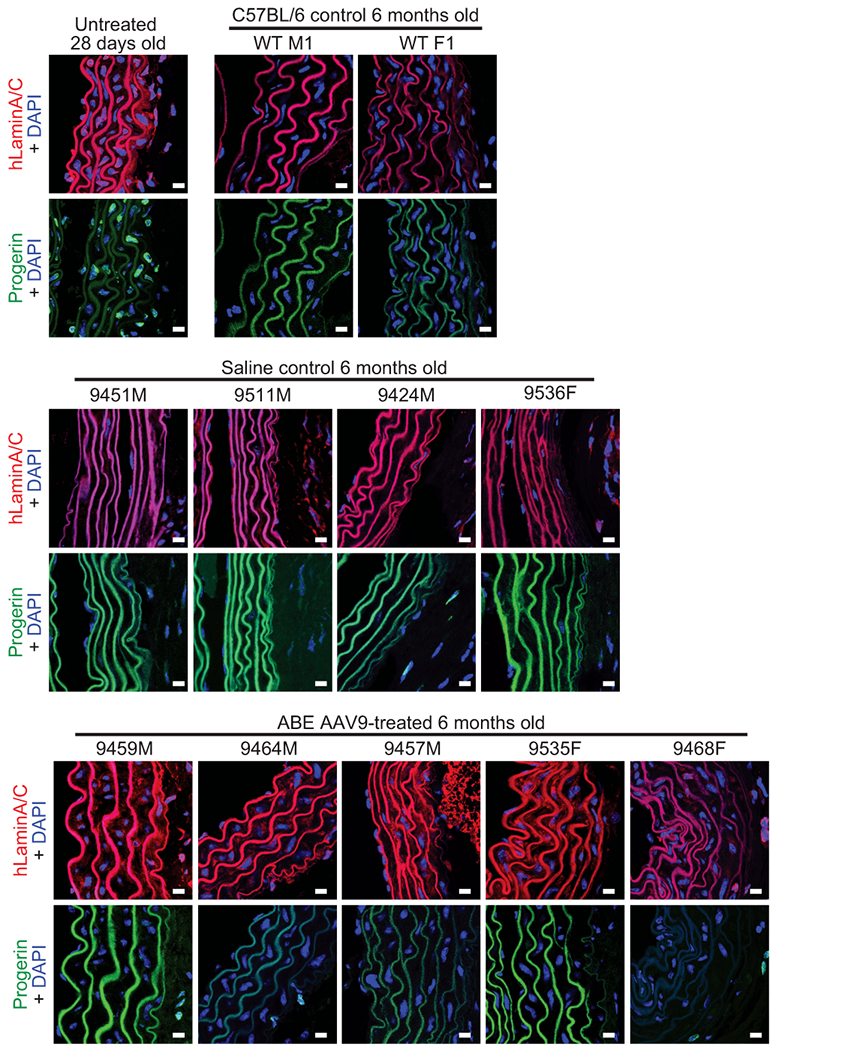

Comment in
-
Base editor repairs mutation found in the premature-ageing syndrome progeria.Nature. 2021 Jan;589(7843):522-524. doi: 10.1038/d41586-020-03573-x. Nature. 2021. PMID: 33408358 No abstract available.
-
Fixing an ageing mutation.Nat Rev Genet. 2021 Mar;22(3):134-135. doi: 10.1038/s41576-021-00327-x. Nat Rev Genet. 2021. PMID: 33437031 No abstract available.
-
In vivo base editing rescues progeria in mice.Nat Rev Drug Discov. 2021 Feb;20(2):98. doi: 10.1038/d41573-021-00013-x. Nat Rev Drug Discov. 2021. PMID: 33441999 No abstract available.
-
Jumping on base editing to repair the diseased cardiovascular system in vivo.Cardiovasc Res. 2021 Mar 21;117(4):e46-e48. doi: 10.1093/cvr/cvab057. Cardiovasc Res. 2021. PMID: 33749783 No abstract available.
-
CRISPR base editor treats premature-aging syndrome.Signal Transduct Target Ther. 2021 Apr 16;6(1):158. doi: 10.1038/s41392-021-00576-6. Signal Transduct Target Ther. 2021. PMID: 33863871 Free PMC article. No abstract available.
References
-
- Gordon LB, Brown WT & Collins FS Hutchinson-Gilford progeria syndrome. GeneReviews (2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
