

Crystal structure of bacterial cytotoxic necrotizing factor CNFY reveals molecular building blocks for intoxication
- PMID: 33410511
- PMCID: PMC7883292
- DOI: 10.15252/embj.2020105202
Crystal structure of bacterial cytotoxic necrotizing factor CNFY reveals molecular building blocks for intoxication
Abstract
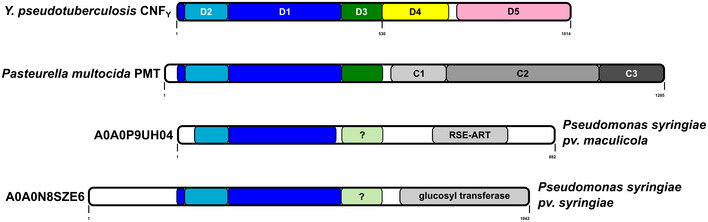
Cytotoxic necrotizing factors (CNFs) are bacterial single-chain exotoxins that modulate cytokinetic/oncogenic and inflammatory processes through activation of host cell Rho GTPases. To achieve this, they are secreted, bind surface receptors to induce endocytosis and translocate a catalytic unit into the cytosol to intoxicate host cells. A three-dimensional structure that provides insight into the underlying mechanisms is still lacking. Here, we determined the crystal structure of full-length Yersinia pseudotuberculosis CNFY . CNFY consists of five domains (D1-D5), and by integrating structural and functional data, we demonstrate that D1-3 act as export and translocation module for the catalytic unit (D4-5) and for a fused β-lactamase reporter protein. We further found that D4, which possesses structural similarity to ADP-ribosyl transferases, but had no equivalent catalytic activity, changed its position to interact extensively with D5 in the crystal structure of the free D4-5 fragment. This liberates D5 from a semi-blocked conformation in full-length CNFY , leading to higher deamidation activity. Finally, we identify CNF translocation modules in several uncharacterized fusion proteins, which suggests their usability as a broad-specificity protein delivery tool.
Keywords: Yersinia; AB-toxin; ADP-ribosyl transferase; CNF; DUF4765.
© 2021 The Authors. Published under the terms of the CC BY NC ND 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
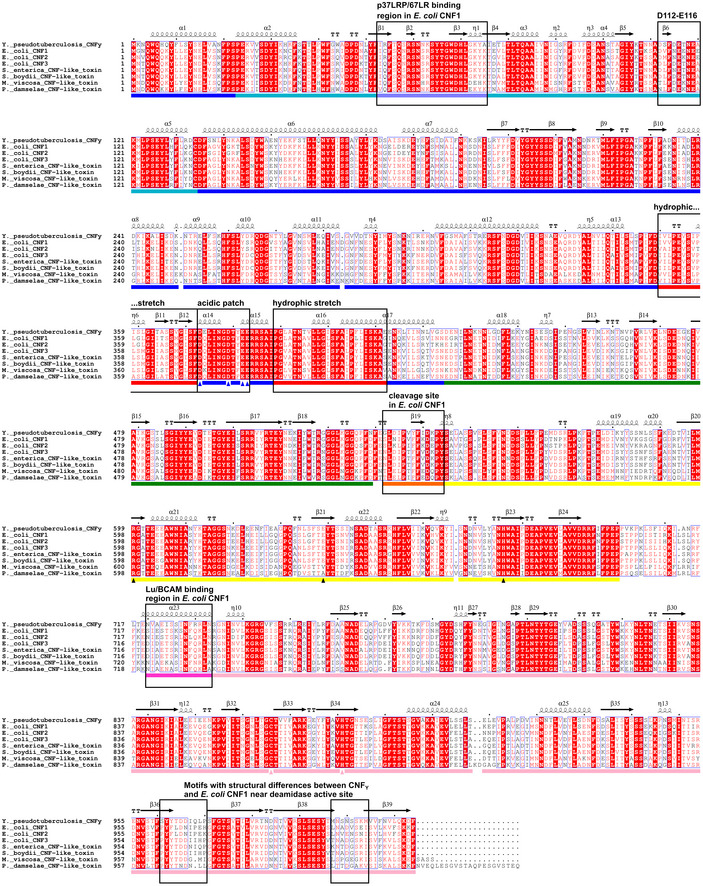
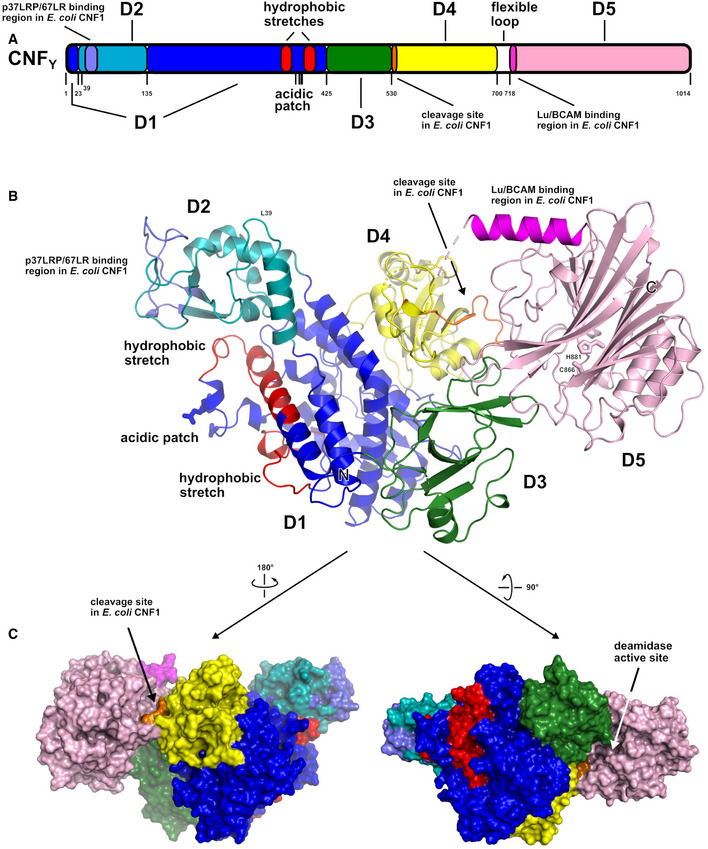
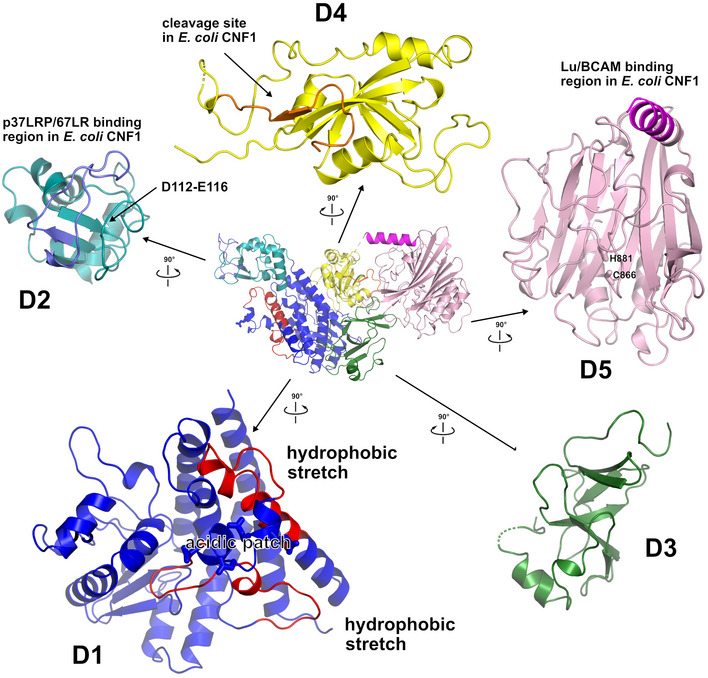
Domain boundaries and sequence motifs mapped to the sequence of CNFY.
Cartoon representation of CNFY, colored according to domain boundaries determined with PiSQRD (Aleksiev et al, 2009). Dark blue: domain D1, cyan: domain D2, dark green: domain D3, yellow: ADP‐ribosyltransferase‐like domain D4, pink: deamidase domain D5. Other colors indicate the position of sequence motifs that have been identified in E. coli CNF1, namely light blue: p37LRP/67LR receptor‐binding motif, red: hydrophobic stretches predicted to form membrane‐inserting α‐helices, orange: cleavage site, magenta: main Lu/BCAM receptor‐binding motif. The positions of N‐ and C‐terminus are indicated by N and C, respectively.
Surface representation of CNFY as seen from two different orientations with respect to (B). Note that the cleavage site between D3 and D4 (orange) as well as the deamidase active site in D5 are partially blocked in the structure of full‐length CNFY. The C‐terminal domain D5 interacts mainly with D3 (610 Å2), which partially hides the catalytic site of D5, but it interacts only weakly with D4 (380 Å2), which itself establishes an extensive interface with D1 (1,390 Å2) by mainly hydrophilic interactions (17 hydrogen bonds and 6 salt bridges).
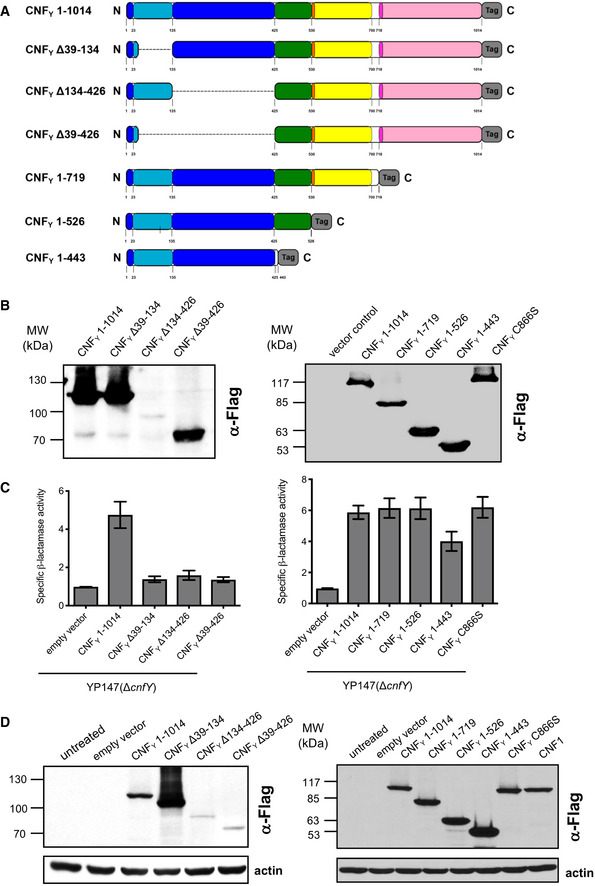
Schematic overview of marker‐tagged CNFY deletion variants.
3xFlag‐tagged CNFY deletion variants were expressed in Y. pseudotuberculosis YP147 (ΔcnfY) from plasmids under control of their own promoter and were detected in whole cell extracts using an anti‐Flag antibody.
To test secretion of the CNFY variants, full‐length CNFY and different N‐ and C‐terminally deleted variants fused to β‐lactamase (TEM) were expressed in Y. pseudotuberculosis YP147 (ΔcnfY). β‐lactamase activity in the culture supernatant was subsequently measured using nitrocefin as substrate. The data represent the mean ± SD of three independent experiments, carried out in triplicates.
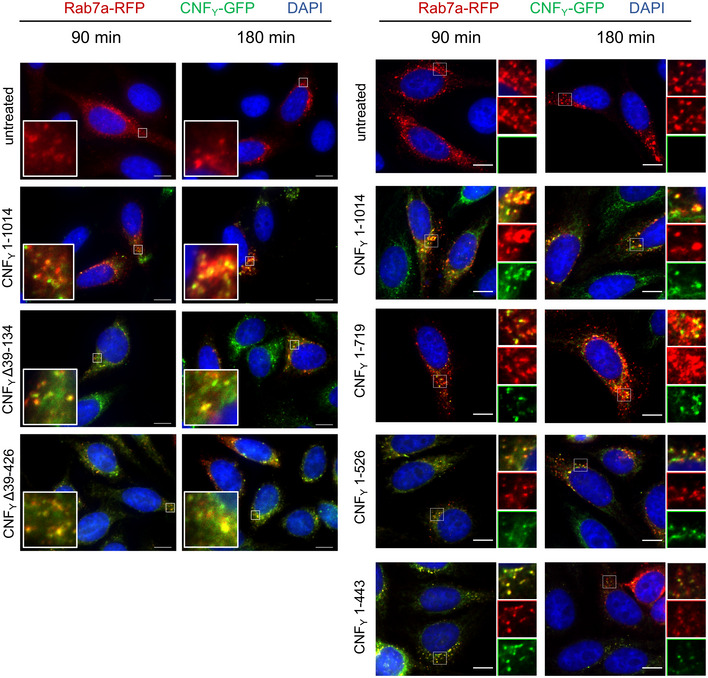
HEp‐2 cells remained untreated or were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing full‐length CNF1, CNFY or the N‐ or C‐terminally deleted toxin variants at 37°C for 4 h. The cells were thoroughly washed, pelleted, lysed and the toxin variants bound to the cells were identified by western blotting using an anti‐Flag antibody.
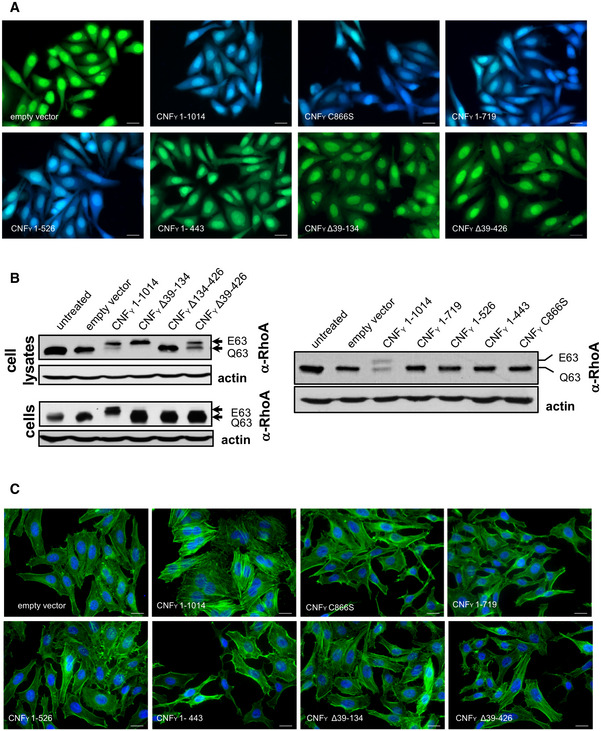
HEp‐2 cells were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing full‐length CNFY or the N‐ or C‐terminally deleted toxin variants fused to β‐lactamase (TEM) at 37°C for 4 h. Cleavage of the reporter dye CCF4‐AM was used to visualize toxin delivery. After cell entry, CCF4‐AM is rapidly converted into the negatively charged form CCF4, which is retained in the cytosol and emits a green fluorescence signal (530 nm). In the presence of translocated β‐lactamase fusion proteins, CCF4‐AM is cleaved, and disruption of FRET results in blue fluorescence (450 nm). Scale bar: 20 µm.
Left upper and right panel: HEp‐2 cells remained untreated or were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing full‐length CNFY or the N‐ or C‐terminally deleted toxin variants for 4 h. Cells were lysed and the deamidation of RhoA was analyzed by the shift of the modified Rho GTPase band in SDS–PAGE gels; left lower panel: HEp‐2 cells were lysed and the cell extracts were incubated with full‐length CNFY or the N‐terminally deleted toxin variants for 4 h. The deamidation of RhoA in the cell extracts was analyzed by the mobility shift of the modified Rho GTPase on SDS–PAGE after detection with anti‐RhoA antibodies.
HEp‐2 cells were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing full‐length CNFY or the N‐ or C‐terminally deleted toxin variants for 24 h. The cell nuclei were stained with DAPI (blue) and the actin cytoskeleton was stained using FITC‐phalloidin (green). The formation of large, multinuclear cells was observed by fluorescence microscopy and the formation of thick actin stress fibers and membrane actin folding were only observed with CNFY‐treated cells. The white scale bar is 40 µm. Cells incubated with extracts of YP147 (ΔcnfY) harboring the empty expression vector were used as negative controls.
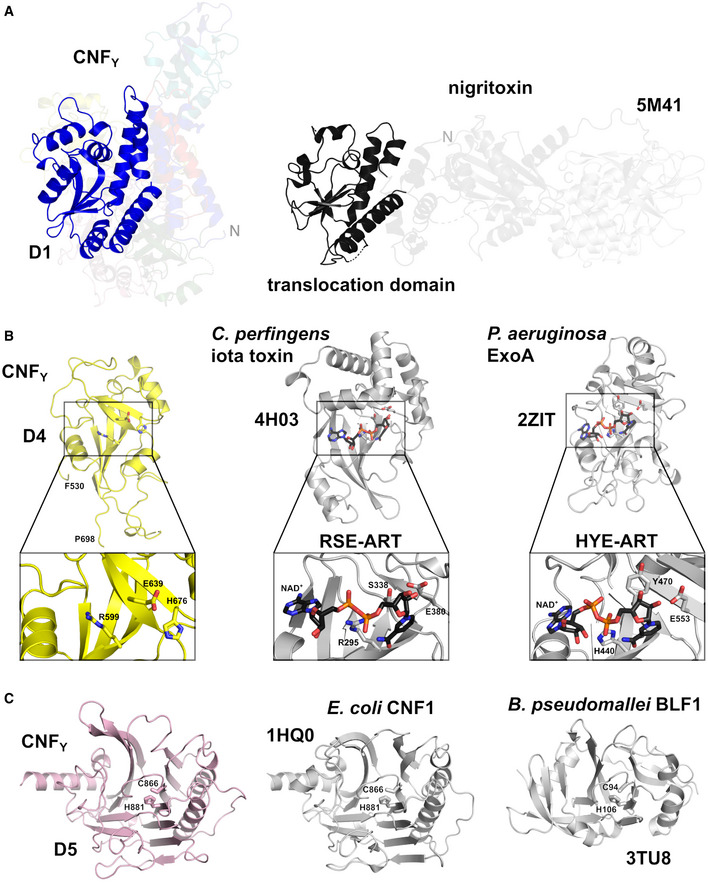
Side‐by‐side comparison of CNFY and nigritoxin. Nigritoxin is a toxin of crustaceans and insects. The translocation domain of nigritoxin [PDB entry 5M41, (Labreuche et al, 2017)] and domain D1 of CNFY show partial structural similarity (highlighted areas). This similarity was identified with DALI (Holm & Rosenström, 2010) which was also used to align both structures.
The ART‐like domain D4 of CNFY. Essential residues of canonical ARTs are not conserved in CNFY (RSE‐ARTs exemplified by C. perfringens iota toxin, PDB entry 4H03 (Tsurumura et al, 2013); HYE‐ARTs exemplified by P. aeruginosa ExoA, PDB entry 2ZIT (Jørgensen et al, 2008); carbon atoms of NAD+ shown in black).
The deamidase domain D5 of CNFY. C866 and H881 form a conserved catalytic dyad also found in the deamidase domain of E. coli CNF1 [PDB entry 1HQ0, (Buetow et al, 2001)] and Burholderia pseudomallei lethal factor BLF1 [PDB entry 3TU8, (Cruz‐Migoni et al, 2011)].
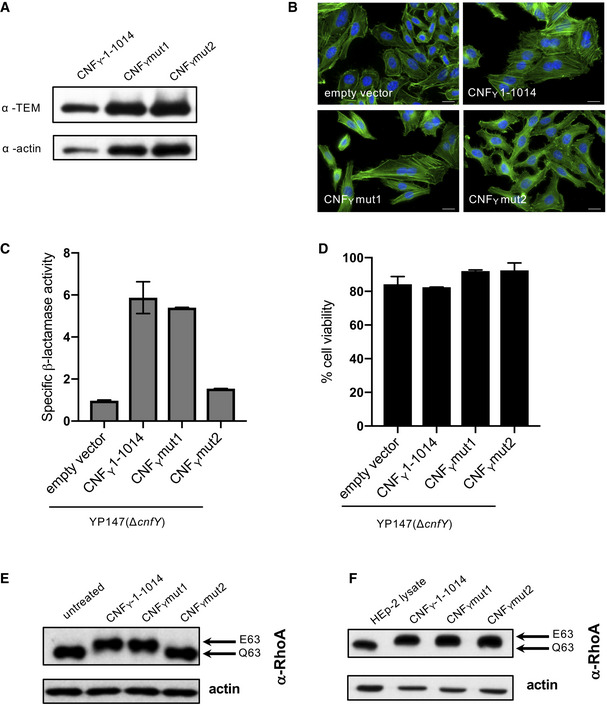
HEp‐2 cells were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing CNFY, the toxin variant mut1: CNFYI535L/P536A/V537G or mut2: CNFYI535L/P536A/V537G/F539L/D541A/K542A fused to TEM or no CNFY protein (empty vector) for 4 h. Cells were lysed and the binding of the different CNFY proteins to HEp‐2 cells was analyzed by immunoblotting.
HEp‐2 cells were incubated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing CNFY, the toxin variant mut1: CNFYI535L/P536A/V537G or mut2: CNFYI535L/P536A/V537G/F539L/D541A/K542A or no CNFY protein (empty vector). The cell nuclei were stained with DAPI (blue) and the actin cytoskeleton was stained using FITC‐phalloidin (green). The results indicated the formation of polynucleated cells and stress fibers only in cells treated with CNFY and CNFYI535L/P536/V537G. The white scale bar is 20 µm.
Nitrocefin (2 mM) was added to the supernatant from 25°C overnight Yersinia cultures expressing the indicated CNFY derivatives to determine β‐lactamase activity by measuring changes in absorbance at 390 nm (yellow) and 486 nm (red). The data represent the mean ± SD of three independent experiments, carried out in triplicates.
The viability of Y. pseudotuberculosis YPIII expressing the indicated CNFY derivatives was assessed in equalized bacterial cultures using the BacTiter‐Glo Microbial Cell Viability Assay kit (Promega). The data represent the mean ± SD of three independent experiments, carried out in triplicates.
HEp‐2 cells treated with 20 µg/ml of whole cell extract of Y. pseudotuberculosis expressing indicated CNFY variants for 4 h were lysed and the deamidation of RhoA was analyzed by the mobility shift of the modified RhoA GTPase detected by immunoblotting.
The activity of the CNFY derivatives was tested by analyzing the deamidation of RhoA in HEp‐2 cell lysates by the mobility shift of the modified GTPase detected by immunoblotting.
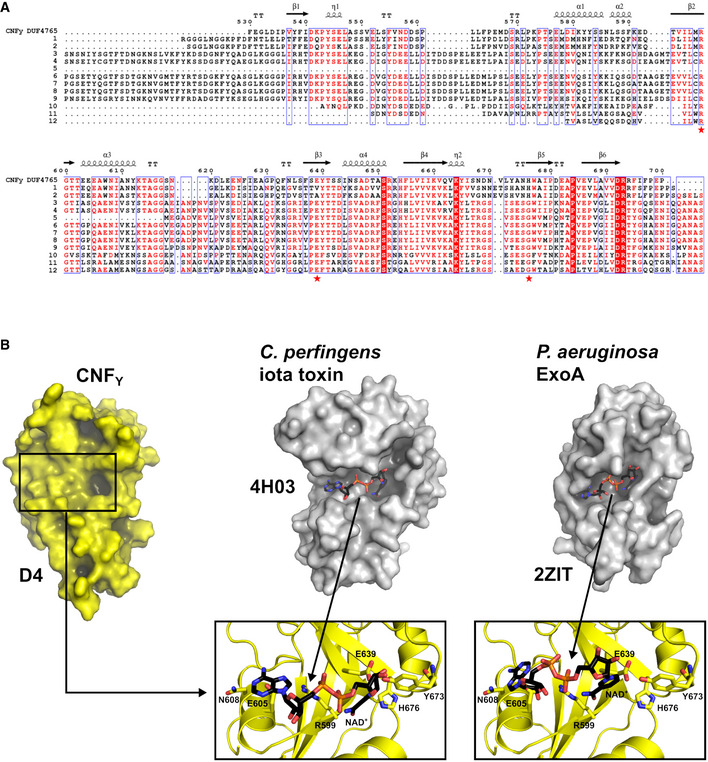
Sequence alignment of representative sequences of the DUF4765 family. Red stars indicate the position of three residues that reside in similar positions as catalytical residues in canonical ART domains (R599, E639, H676 in CNFY). The aligned sequences are 1: M. viscosa, CNF1, 55% sequence similarity to domain 4 of CNFY (UniProt‐ID: W6AYD9), 2: P. damselae, CNF1, 55% seq. similarity (UniProt‐ID: D0Z517), 3: Bacteriophage Stx2a_WGPS2, EspN, 23% seq. similarity (UniProt‐ID: A0A0P0ZCL8), 4: C. rodentium, EspN2‐2, 23% seq. similarity (UniProt‐ID: D2TQZ7), 5: S. bongori, EspN, 24% seq. similarity (UniProt‐ID: A0A3S5D9E8), 6: S. enterica subsp. arizonae, uncharacterized protein, 23% seq. similarity (UniProt‐ID: A9MRQ7), 7: S. enterica subsp. arizonae, uncharacterized protein, 24% seq. similarity (UniProt‐ID: A9MQT6), 8: S. enterica subsp. arizonae, EspN, 24% seq. similarity (UniProt‐ID: A0A2X4TBS3), 9: E. coli O157:H7, EspN, 26% seq. similarity (UniProt‐ID: A0A0H3JD33), 10: P. temperata, uncharacterized protein, 18% seq. similarity (UniProt‐ID: T0QEQ3), 11: Streptomyces sp. H‐KF8, uncharacterized protein, 20% seq. similarity (UniProt‐ID: A0A1A5P6M0), 12: S. xinghaiensis, uncharacterized protein, 22% seq. similarity (UniProt‐ID: A0A420VA44). The sequence alignment has been performed with PROMALS3D (Pei et al, 2008) and was rendered with ESPript (Gouet et al, 2003).
Comparison of the ART‐like domain D4 of CNFY with canonical ART domains. Note that the hypothetical NAD+ binding site of D4 is shallower than that of the other two domains and that the NAD+ molecules found in these examples would clash with residues of D4 when bound in the same conformation.
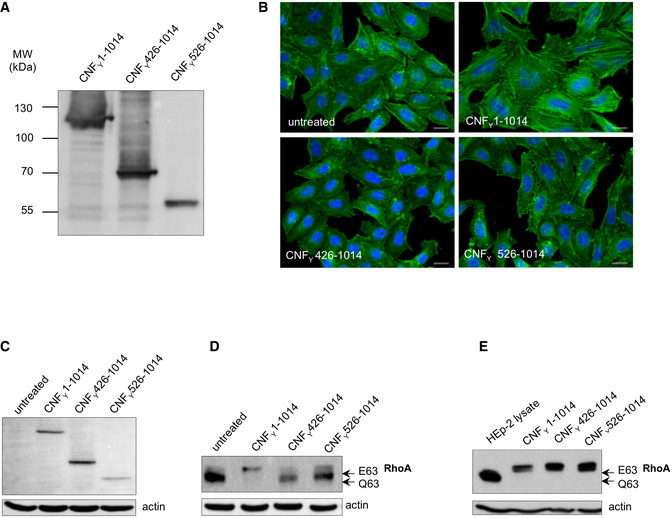
The expression of purified recombinant CNFY N‐terminal domains D3–5 (CNFY 426–1,014) or D4–5 (CNFY 526–1,014).
HEp‐2 cells remain untreated or were incubated with 500 nM purified full‐length CNFY, domains D3–5 (CNFY 426–1,014) or D4–5 (CNFY 526–1,014) for 24 h. The formation of large, multinuclear cells was observed by fluorescence microscopy. The cell nuclei were stained with DAPI (blue) and the actin cytoskeleton was stained using FITC‐Phalloidin (green). The white scale bar is 20 µm.
Binding of purified full‐length CNFY, domains D3–5 (CNFY 426–1,014) or D4–5 (CNFY 526–1014) to HEp‐2 cells was analyzed by immunoblotting after HEp‐2 cells were incubated with 500 nM of the purified toxin or toxin fragments for 4 h.
HEp‐2 cells treated with 500 nM purified full‐length CNFY, protein fragments D3–5 (CNFY 426–1,014) or D4–5 (CNFY 526–1,014) were lysed and the deamidation of RhoA was analyzed by the mobility shift of the modified GTPase detected by immunoblotting.
The activity of purified CNFY and the protein fragments D3–5 (CNFY 426–1,014) or D4–5 (CNFY 526–1,014) was tested by analyzing the deamidation of RhoA in HEp‐2 cell lysates by the mobility shift of the modified GTPase detected by immunoblotting.
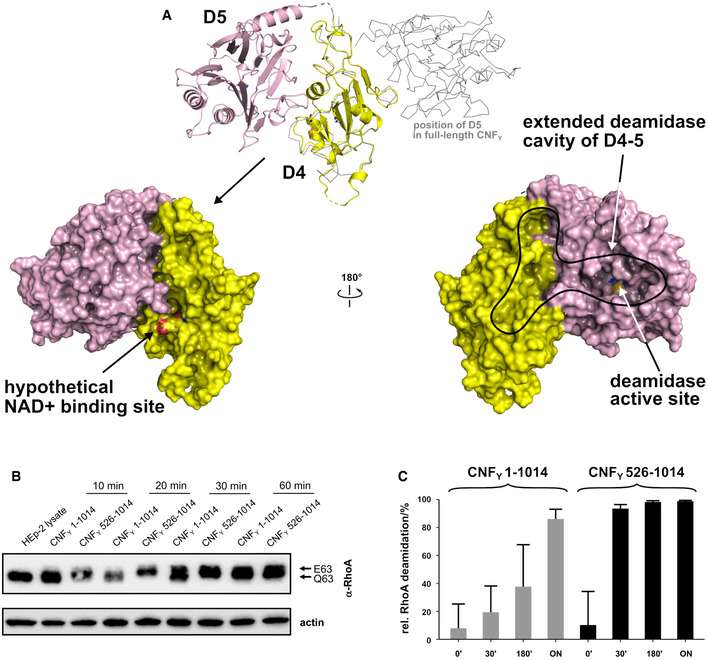
Crystal structure of the free D4–5 subunit. Note the different relative orientations of domains D4 and D5 with respect to the structure of full‐length CNFY (top, thin gray lines). The domain D4 forms a large interface area (1,100 Å2) with the catalytic domain D5 involving several polar interactions (8 hydrogen bonds and 8 salt bridges), whereby the active crevice is extended and fully solvent‐exposed as can be seen in the right surface plot at the bottom of the panel. The hypothetical NAD+ binding site of the ART‐like D4 domain is located on the opposite face (left surface plot). Note that the deamidase active site of domain D5, unlike in the full‐length structure (Fig 1), is fully accessible and that its extended shape is also determined by domain D4.
Comparative analysis of RhoA activation in HEp‐2 cell lysate by CNFY and the recombinant D4–5 protein. Purified CNFY or the D4–5 fragment (1 µM) was added to extracts of HEp‐2 cells and incubated for 10, 20, 30, or 60 min. Deamidation of RhoA was analyzed by the shift of the modified Rho GTPase band in SDS–PAGE gels after detection with anti‐RhoA antibodies.
Comparative analysis of recombinant RhoA deamidation by CNFY and the D4–5 fragment. Recombinant RhoA was incubated with purified CNFY or the D4–5 fragment and samples were separated by SDS–PAGE after the indicated times before subjecting to trypsin digestion and quantification of deamidation of Q63 by mass spectrometry. Error bars represent standard deviations of triplicate measurements.
Similar articles
-
The Cytotoxic Necrotizing Factors (CNFs)-A Family of Rho GTPase-Activating Bacterial Exotoxins.Toxins (Basel). 2021 Dec 15;13(12):901. doi: 10.3390/toxins13120901. Toxins (Basel). 2021. PMID: 34941738 Free PMC article. Review.
-
The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA.J Biol Chem. 2004 Apr 16;279(16):16026-32. doi: 10.1074/jbc.M313556200. Epub 2004 Feb 3. J Biol Chem. 2004. PMID: 14761941
-
The cytotoxic necrotizing factor of Yersinia pseudotuberculosis (CNFY) enhances inflammation and Yop delivery during infection by activation of Rho GTPases.PLoS Pathog. 2013;9(11):e1003746. doi: 10.1371/journal.ppat.1003746. Epub 2013 Nov 7. PLoS Pathog. 2013. PMID: 24244167 Free PMC article.
-
Modular domain swapping among the bacterial cytotoxic necrotizing factor (CNF) family for efficient cargo delivery into mammalian cells.J Biol Chem. 2018 Mar 9;293(10):3860-3870. doi: 10.1074/jbc.RA117.001381. Epub 2018 Jan 25. J Biol Chem. 2018. PMID: 29371399 Free PMC article.
-
Cytotoxic Necrotizing Factors (CNFs)-A Growing Toxin Family.Toxins (Basel). 2010 Jan;2(1):116-27. doi: 10.3390/toxins2010116. Epub 2011 Apr 8. Toxins (Basel). 2010. PMID: 22069550 Free PMC article. Review.
Cited by
-
The cnf1 gene is associated with an expanding Escherichia coli ST131 H30Rx/C2 subclade and confers a competitive advantage for gut colonization.Gut Microbes. 2022 Jan-Dec;14(1):2121577. doi: 10.1080/19490976.2022.2121577. Gut Microbes. 2022. PMID: 36154446 Free PMC article.
-
Bacterial AB toxins and host-microbe interactions.Adv Microb Physiol. 2022;81:67-109. doi: 10.1016/bs.ampbs.2022.06.002. Epub 2022 Jul 18. Adv Microb Physiol. 2022. PMID: 36167443 Free PMC article. Review.
-
Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS.Genes (Basel). 2024 Sep 26;15(10):1250. doi: 10.3390/genes15101250. Genes (Basel). 2024. PMID: 39457374 Free PMC article.
-
The Cytotoxic Necrotizing Factors (CNFs)-A Family of Rho GTPase-Activating Bacterial Exotoxins.Toxins (Basel). 2021 Dec 15;13(12):901. doi: 10.3390/toxins13120901. Toxins (Basel). 2021. PMID: 34941738 Free PMC article. Review.
-
Unconventional structure and mechanisms for membrane interaction and translocation of the NF-κB-targeting toxin AIP56.Nat Commun. 2023 Nov 16;14(1):7431. doi: 10.1038/s41467-023-43054-z. Nat Commun. 2023. PMID: 37973928 Free PMC article.
References
-
- Aleksiev T, Potestio R, Pontiggia F, Cozzini S, Micheletti C (2009) PiSQRD: a web server for decomposing proteins into quasi‐rigid dynamical domains. Bioinformatics 25: 2743–2744 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
