

CDK2-Mediated Upregulation of TNFα as a Mechanism of Selective Cytotoxicity in Acute Leukemia
- PMID: 33414171
- PMCID: PMC8137540
- DOI: 10.1158/0008-5472.CAN-20-1504
CDK2-Mediated Upregulation of TNFα as a Mechanism of Selective Cytotoxicity in Acute Leukemia
Abstract
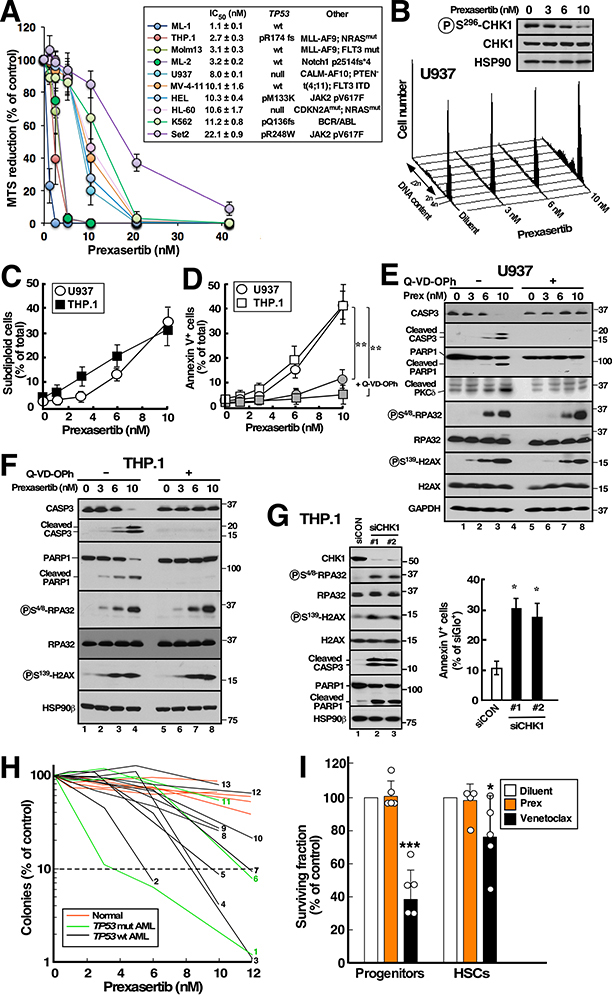
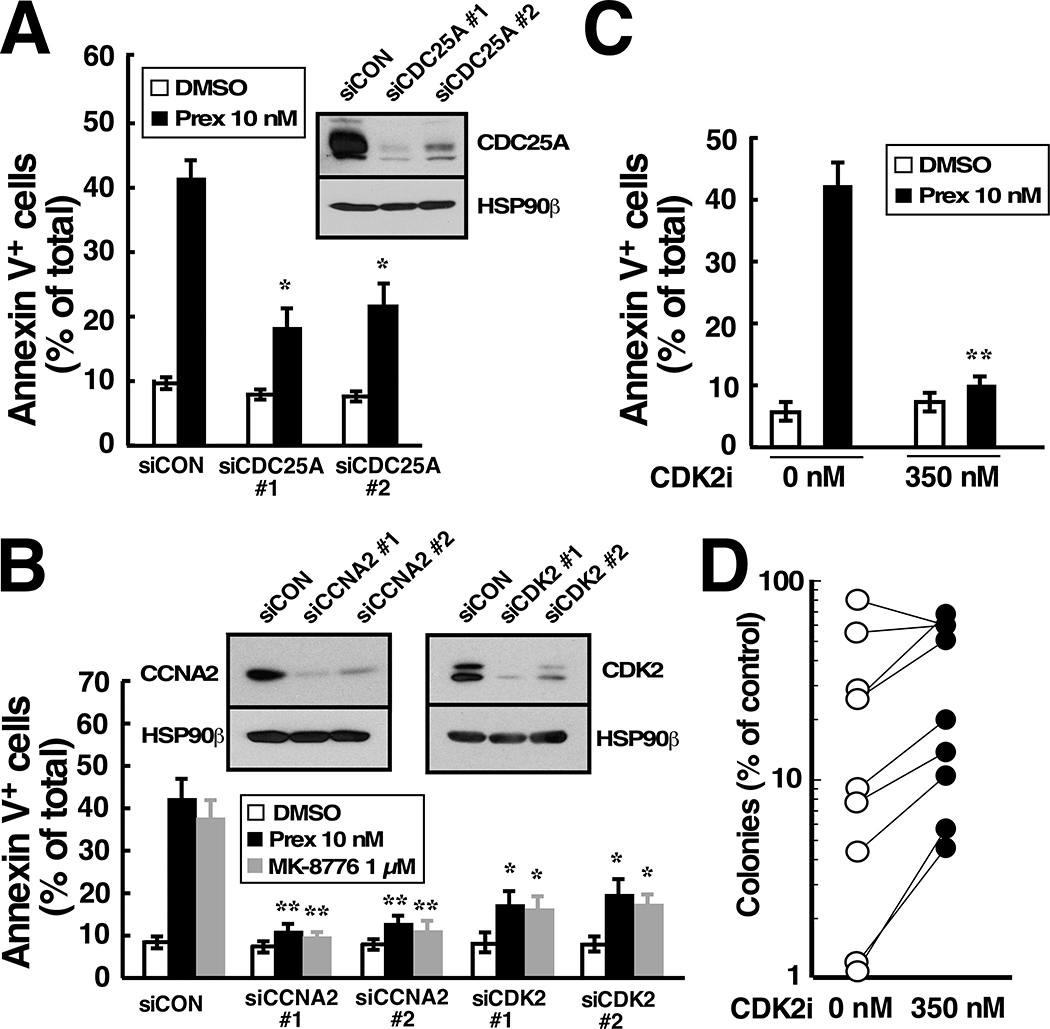
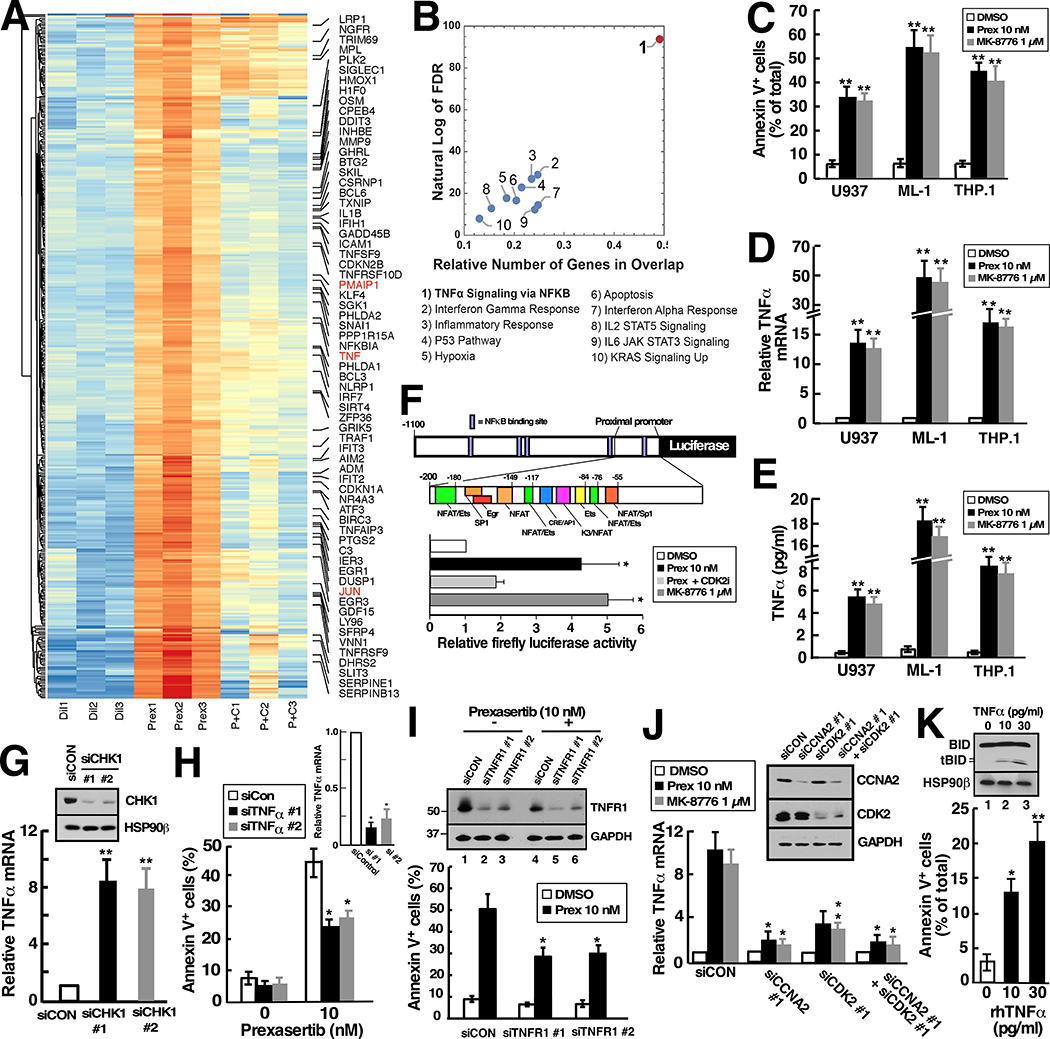
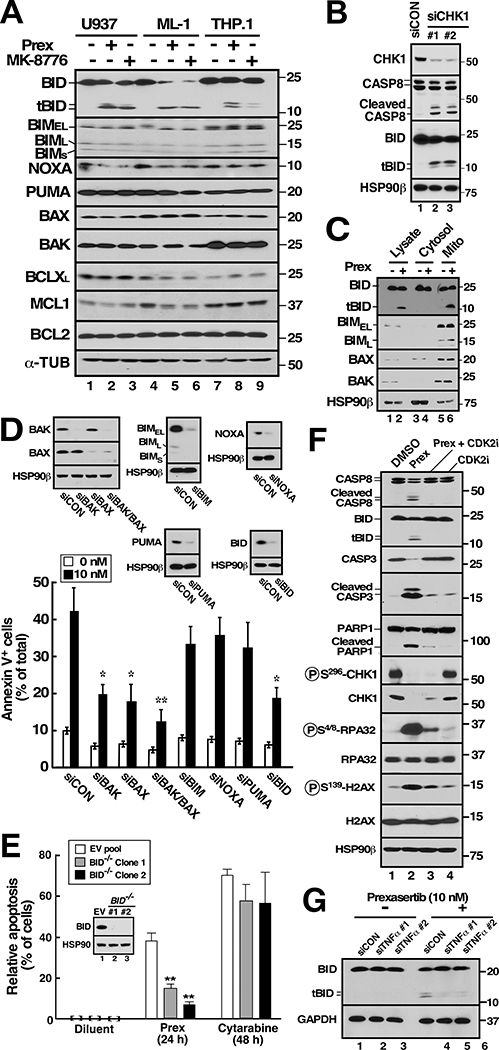
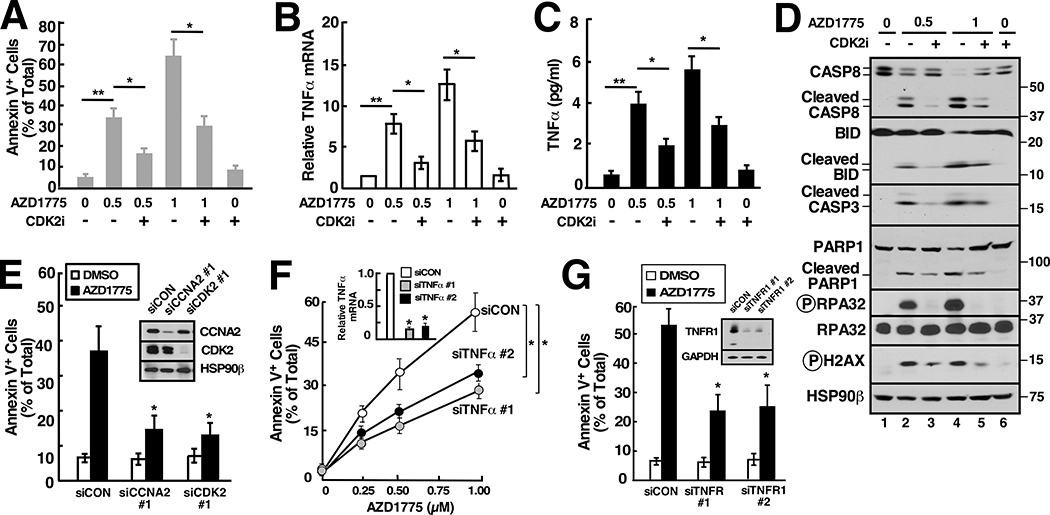
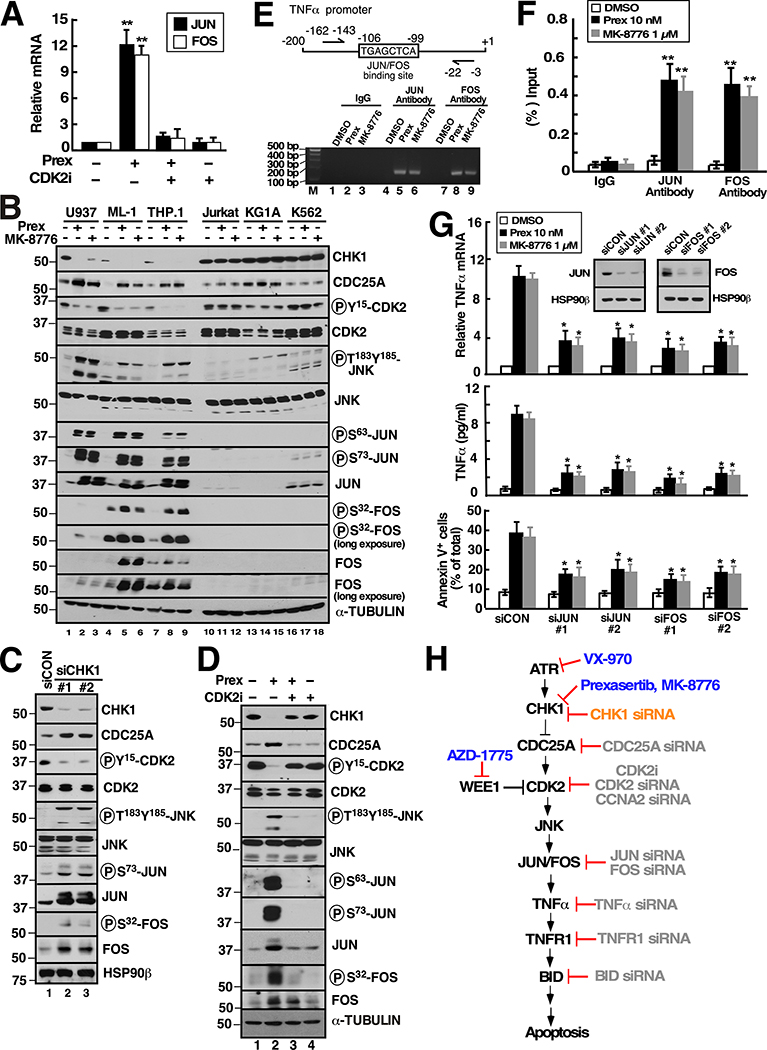
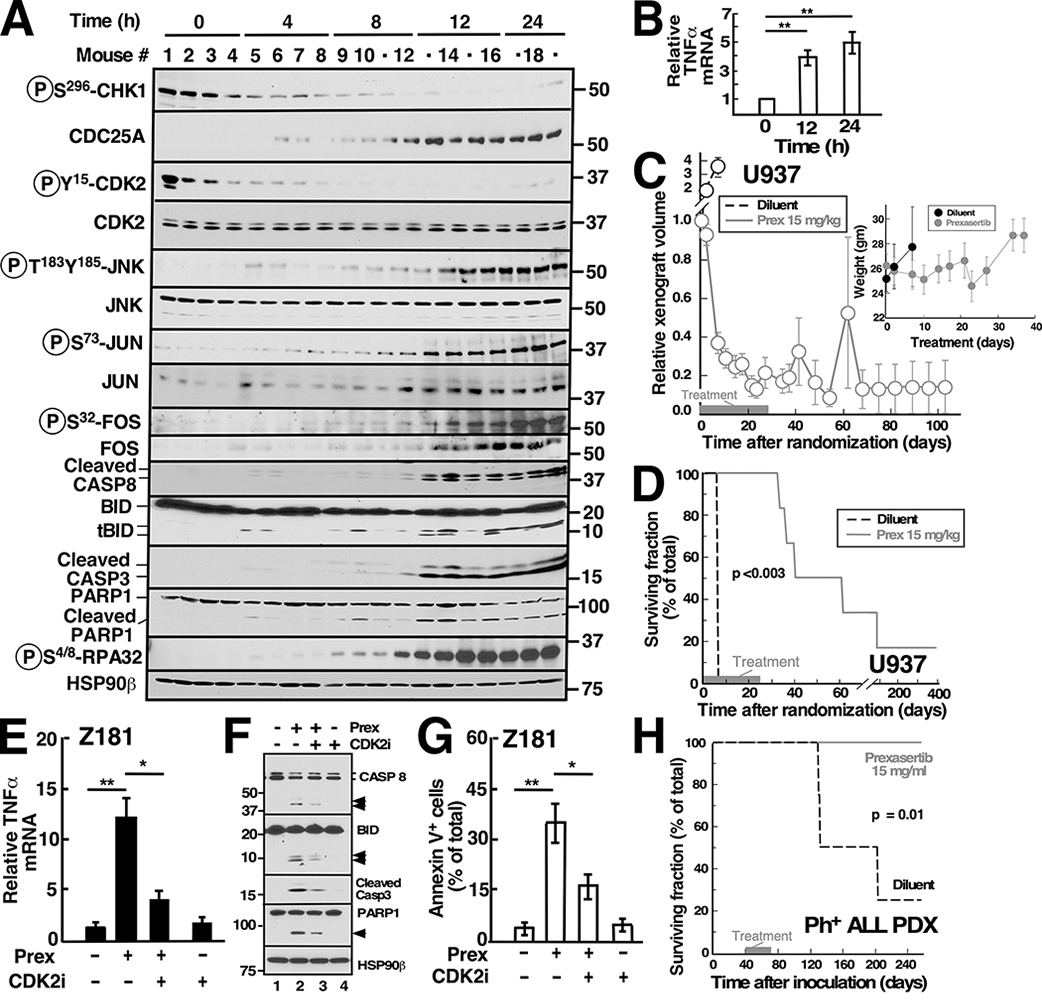
Although inhibitors of the kinases CHK1, ATR, and WEE1 are undergoing clinical testing, it remains unclear how these three classes of agents kill susceptible cells and whether they utilize the same cytotoxic mechanism. Here we observed that CHK1 inhibition induces apoptosis in a subset of acute leukemia cell lines in vitro, including TP53-null acute myeloid leukemia (AML) and BCR/ABL-positive acute lymphoid leukemia (ALL), and inhibits leukemic colony formation in clinical AML samples ex vivo. In further studies, downregulation or inhibition of CHK1 triggered signaling in sensitive human acute leukemia cell lines that involved CDK2 activation followed by AP1-dependent TNF transactivation, TNFα production, and engagement of a TNFR1- and BID-dependent apoptotic pathway. AML lines that were intrinsically resistant to CHK1 inhibition exhibited high CHK1 expression and were sensitized by CHK1 downregulation. Signaling through this same CDK2-AP1-TNF cytotoxic pathway was also initiated by ATR or WEE1 inhibitors in vitro and during CHK1 inhibitor treatment of AML xenografts in vivo. Collectively, these observations not only identify new contributors to the antileukemic cell action of CHK1, ATR, and WEE1 inhibitors, but also delineate a previously undescribed pathway leading from aberrant CDK2 activation to death ligand-induced killing that can potentially be exploited for acute leukemia treatment. SIGNIFICANCE: This study demonstrates that replication checkpoint inhibitors can kill AML cells through a pathway involving AP1-mediated TNF gene activation and subsequent TP53-independent, TNFα-induced apoptosis, which can potentially be exploited clinically.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures
Similar articles
-
CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells.J Hematol Oncol. 2014 Aug 1;7:53. doi: 10.1186/s13045-014-0053-9. J Hematol Oncol. 2014. PMID: 25084614 Free PMC article.
-
CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo.Haematologica. 2014 Apr;99(4):688-96. doi: 10.3324/haematol.2013.093187. Epub 2013 Oct 31. Haematologica. 2014. PMID: 24179152 Free PMC article.
-
Chidamide increases the sensitivity of refractory or relapsed acute myeloid leukemia cells to anthracyclines via regulation of the HDAC3 -AKT-P21-CDK2 signaling pathway.J Exp Clin Cancer Res. 2020 Dec 9;39(1):278. doi: 10.1186/s13046-020-01792-8. J Exp Clin Cancer Res. 2020. PMID: 33298132 Free PMC article.
-
[Biological properties and sensitivity to induction therapy of differentiated cells expressing atypical immunophenotype in acute leukemia of children].Folia Med Cracov. 2001;42(3):5-80. Folia Med Cracov. 2001. PMID: 12353422 Review. Polish.
-
Mitochondria and Acute Leukemia: A Clinician's Perspective.Int J Mol Sci. 2024 Sep 7;25(17):9704. doi: 10.3390/ijms25179704. Int J Mol Sci. 2024. PMID: 39273651 Free PMC article. Review.
Cited by
-
Inhibiting WEE1 and IKK-RELA Crosstalk Overcomes TNFα Resistance in Head and Neck Cancers.Mol Cancer Res. 2022 Jun 3;20(6):867-882. doi: 10.1158/1541-7786.MCR-21-0624. Mol Cancer Res. 2022. PMID: 35176168 Free PMC article.
-
Proapoptotic activity of JNK-sensitive BH3-only proteins underpins ovarian cancer response to replication checkpoint inhibitors.Mol Cancer. 2024 Oct 7;23(1):224. doi: 10.1186/s12943-024-02125-5. Mol Cancer. 2024. PMID: 39375715 Free PMC article.
-
KIF11 serves as a cell cycle mediator in childhood acute lymphoblastic leukemia.J Cancer Res Clin Oncol. 2023 Nov;149(17):15609-15622. doi: 10.1007/s00432-023-05240-w. Epub 2023 Sep 1. J Cancer Res Clin Oncol. 2023. PMID: 37656243 Free PMC article.
-
Cyclin Y interacts with Chk1 to activate RRM2/STAT3 signaling and promotes radioresistance in non-small cell lung cancer.Int J Biol Sci. 2025 Feb 18;21(5):1999-2011. doi: 10.7150/ijbs.106925. eCollection 2025. Int J Biol Sci. 2025. PMID: 40083692 Free PMC article.
References
-
- Ubhi T, Brown GW. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res 2019;79(8):1730–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
