

Differences in TCR repertoire and T cell activation underlie the divergent outcomes of antitumor immune responses in tumor-eradicating versus tumor-progressing hosts
- PMID: 33414263
- PMCID: PMC7797305
- DOI: 10.1136/jitc-2020-001615
Differences in TCR repertoire and T cell activation underlie the divergent outcomes of antitumor immune responses in tumor-eradicating versus tumor-progressing hosts
Abstract
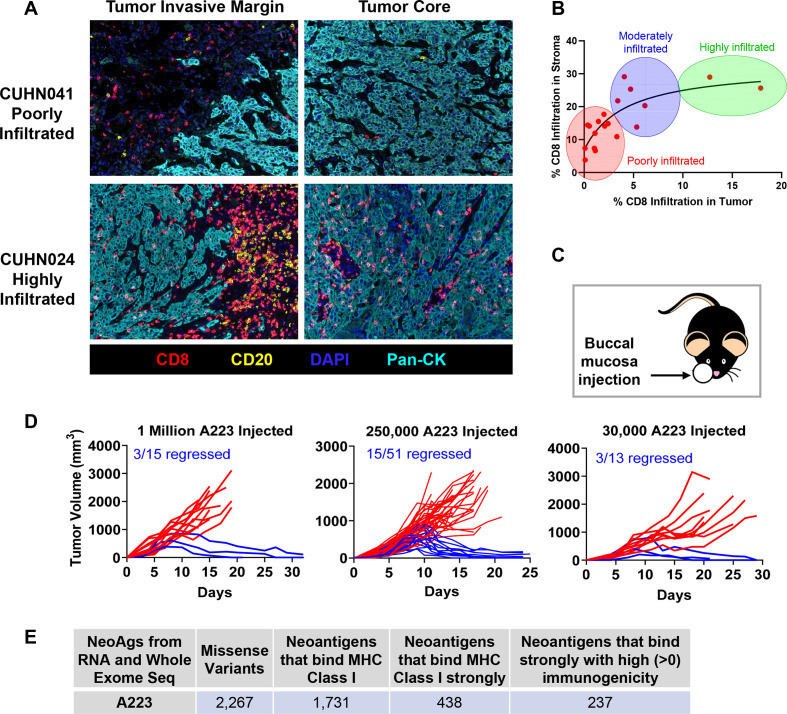
Background: Antitumor immunity is highly heterogeneous between individuals; however, underlying mechanisms remain elusive, despite their potential to improve personalized cancer immunotherapy. Head and neck squamous cell carcinomas (HNSCCs) vary significantly in immune infiltration and therapeutic responses between patients, demanding a mouse model with appropriate heterogeneity to investigate mechanistic differences.
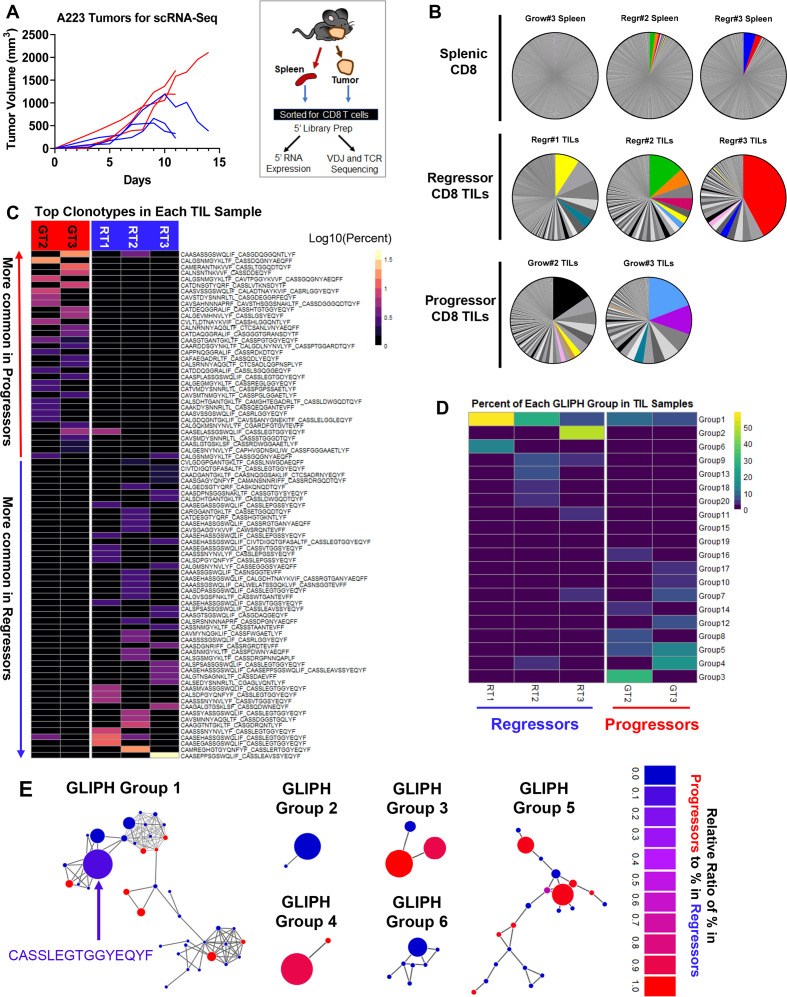
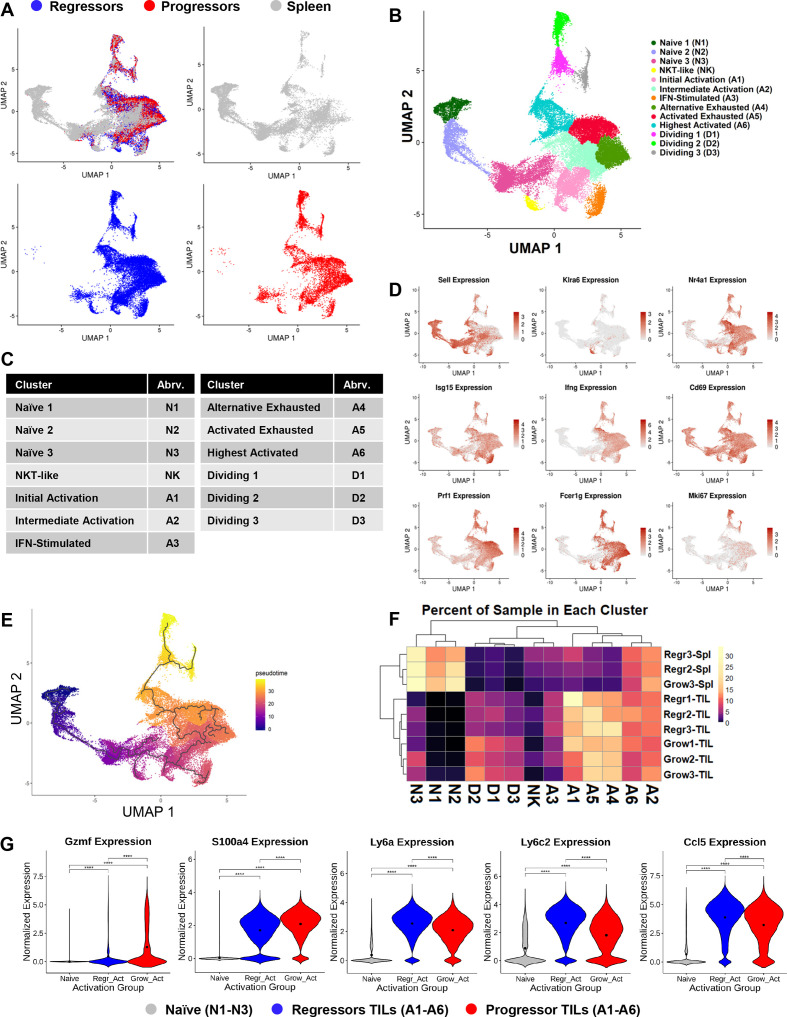
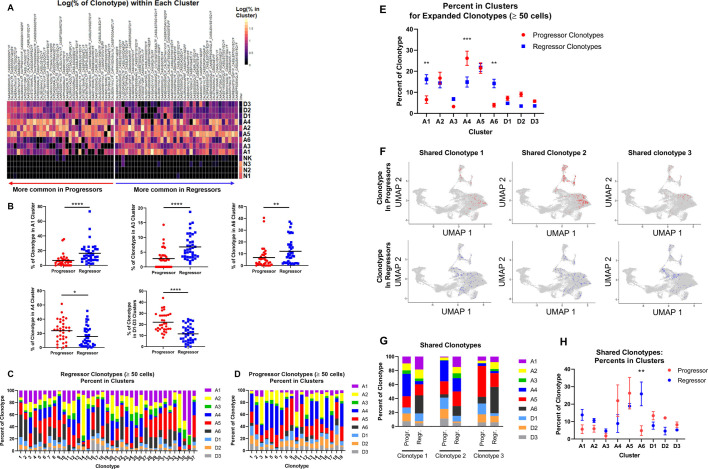
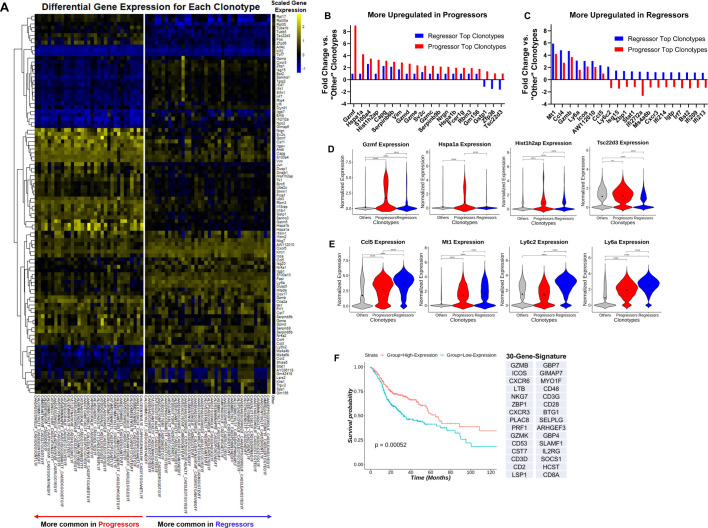
Methods: We developed a unique HNSCC mouse model to investigate underlying mechanisms of heterogeneous antitumor immunity. This model system may provide a better control for tumor-intrinsic and host-genetic variables, thereby uncovering the contribution of the adaptive immunity to tumor eradication. We employed single-cell T-cell receptor (TCR) sequencing coupled with single-cell RNA sequencing to identify the difference in TCR repertoire of CD8 tumor-infiltrating lymphocytes (TILs) and the unique activation states linked with different TCR clonotypes.
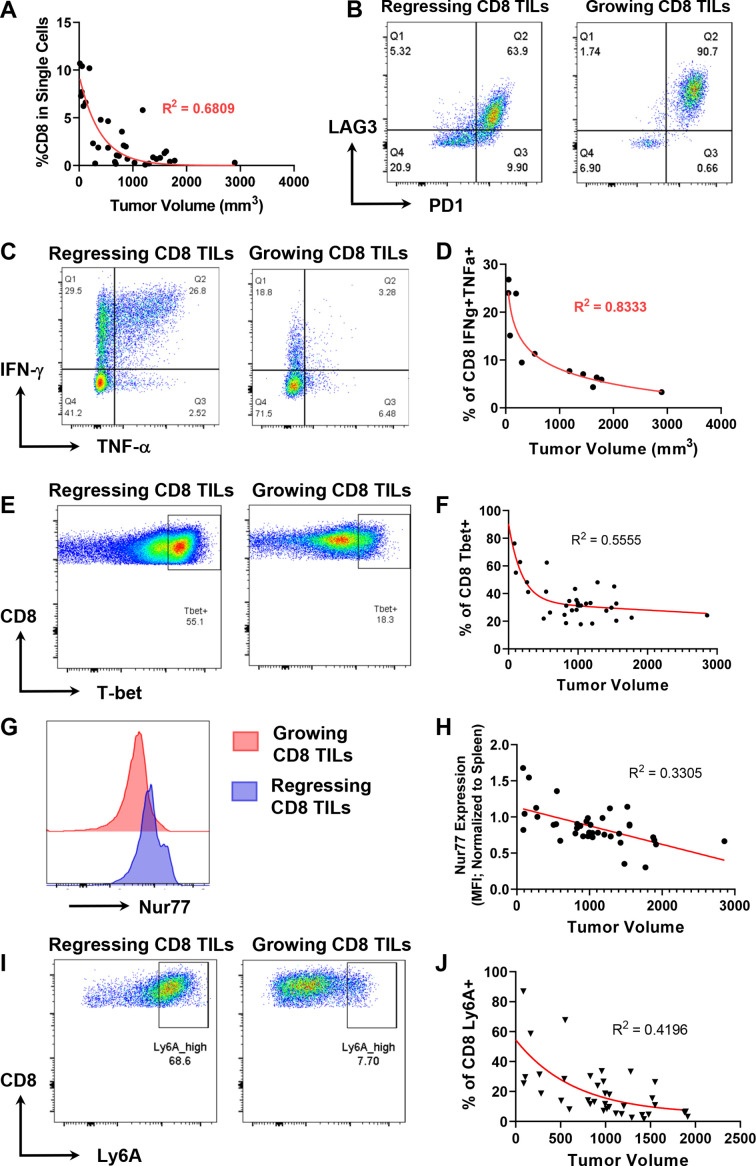
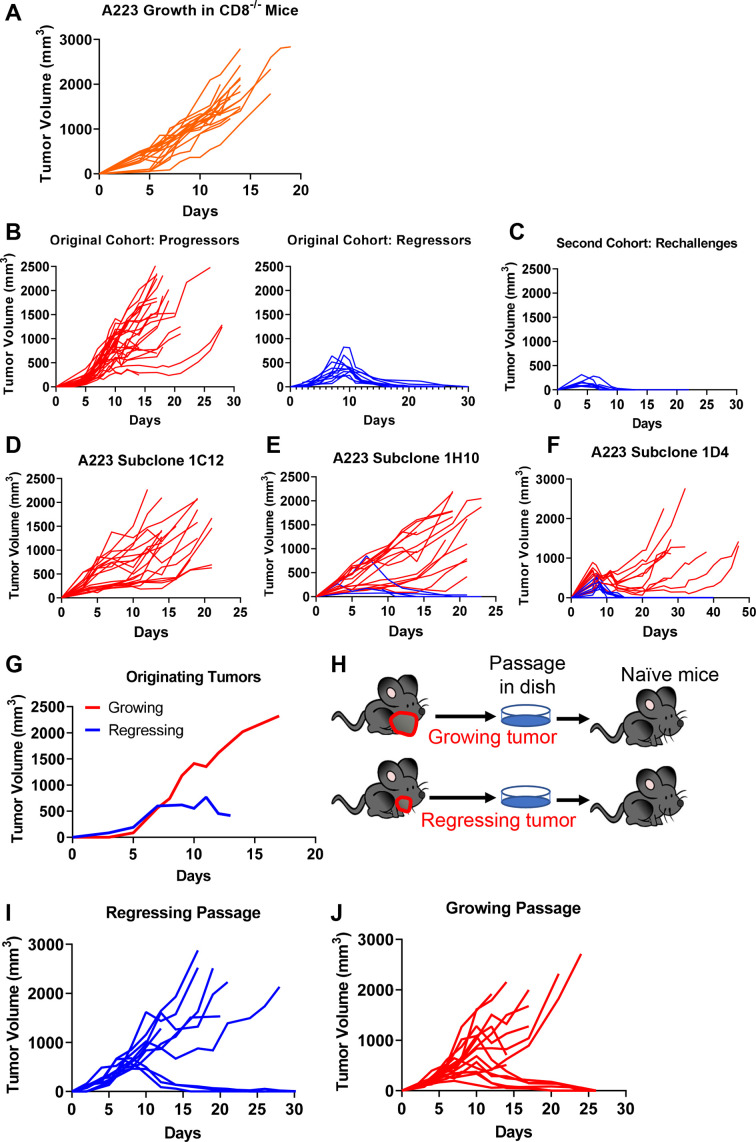
Results: We discovered that genetically identical wild-type recipient mice responded heterogeneously to the same squamous cell carcinoma tumors orthotopically transplanted into the buccal mucosa. While tumors initially grew in 100% of recipients and most developed aggressive tumors, ~25% of recipients reproducibly eradicated tumors without intervention. Heterogeneous antitumor responses were dependent on CD8 T cells. Consistently, CD8 TILs in regressing tumors were significantly increased and more activated. Single-cell TCR-sequencing revealed that CD8 TILs from both growing and regressing tumors displayed evidence of clonal expansion compared with splenic controls. However, top TCR clonotypes and TCR specificity groups appear to be mutually exclusive between regressing and growing TILs. Furthermore, many TCRα/TCRβ sequences only occur in one recipient. By coupling single-cell transcriptomic analysis with unique TCR clonotypes, we found that top TCR clonotypes clustered in distinct activation states in regressing versus growing TILs. Intriguingly, the few TCR clonotypes shared between regressors and progressors differed greatly in their activation states, suggesting a more dominant influence from tumor microenvironment than TCR itself on T cell activation status.
Conclusions: We reveal that intrinsic differences in the TCR repertoire of TILs and their different transcriptional trajectories may underlie the heterogeneous antitumor immune responses in different hosts. We suggest that antitumor immune responses are highly individualized and different hosts employ different TCR specificities against the same tumors, which may have important implications for developing personalized cancer immunotherapy.
Keywords: antigen; head and neck neoplasms; immune evation; immunologic techniques; lymphocytes; receptors; tumor-infiltrating.
© Author(s) (or their employer(s)) 2021. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Divergent outcomes of anti-PD-L1 treatment coupled with host-intrinsic differences in TCR repertoire and distinct T cell activation states in responding versus non-responding tumors.Front Immunol. 2022 Oct 18;13:992630. doi: 10.3389/fimmu.2022.992630. eCollection 2022. Front Immunol. 2022. PMID: 36330507 Free PMC article.
-
Host-specific differences in top-expanded TCR clonotypes correlate with divergent outcomes of anti-PD-L1 treatment in responders versus non-responders.Front Immunol. 2023 Mar 27;14:1100520. doi: 10.3389/fimmu.2023.1100520. eCollection 2023. Front Immunol. 2023. PMID: 37051229 Free PMC article.
-
Differential tumor immune microenvironment coupled with tumor progression or tumor eradication in HPV-antigen expressing squamous cell carcinoma (SCC) models.Front Immunol. 2024 Jul 11;15:1405318. doi: 10.3389/fimmu.2024.1405318. eCollection 2024. Front Immunol. 2024. PMID: 39055715 Free PMC article.
-
Unravelling the heterogeneity and dynamic relationships of tumor-infiltrating T cells by single-cell RNA sequencing analysis.J Leukoc Biol. 2020 Jun;107(6):917-932. doi: 10.1002/JLB.6MR0320-234R. Epub 2020 Apr 9. J Leukoc Biol. 2020. PMID: 32272497 Free PMC article. Review.
-
Effector, Memory, and Dysfunctional CD8(+) T Cell Fates in the Antitumor Immune Response.J Immunol Res. 2016;2016:8941260. doi: 10.1155/2016/8941260. Epub 2016 May 22. J Immunol Res. 2016. PMID: 27314056 Free PMC article. Review.
Cited by
-
Tumor microenvironment in non-melanoma skin cancer resistance to photodynamic therapy.Front Oncol. 2022 Oct 21;12:970279. doi: 10.3389/fonc.2022.970279. eCollection 2022. Front Oncol. 2022. PMID: 36338755 Free PMC article. Review.
-
Differential responses to immune checkpoint inhibitor dictated by pre-existing differential immune profiles in squamous cell carcinomas caused by same initial oncogenic drivers.J Exp Clin Cancer Res. 2022 Apr 2;41(1):123. doi: 10.1186/s13046-022-02337-x. J Exp Clin Cancer Res. 2022. PMID: 35366939 Free PMC article.
-
T-cell repertoire diversity: friend or foe for protective antitumor response?J Exp Clin Cancer Res. 2022 Dec 22;41(1):356. doi: 10.1186/s13046-022-02566-0. J Exp Clin Cancer Res. 2022. PMID: 36550555 Free PMC article. Review.
-
Monitoring Immune Cell Function Through Optical Imaging: a Review Highlighting Transgenic Mouse Models.Mol Imaging Biol. 2022 Apr;24(2):250-263. doi: 10.1007/s11307-021-01662-5. Epub 2021 Nov 4. Mol Imaging Biol. 2022. PMID: 34735680 Free PMC article. Review.
-
Progress in immunotherapy for neuroendocrine neoplasm of the digestive system.World J Gastroenterol. 2023 Jul 14;29(26):4174-4185. doi: 10.3748/wjg.v29.i26.4174. World J Gastroenterol. 2023. PMID: 37475845 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials